The results of standardization and quality control of the drug “Bralecord” solution for infusions are given, according to such quality indicators as: description, authenticity, transparency, color, pH, mechanical inclusions, impurities, osmolality.
## I. INTRODUCTION
Processes of globalization of the pharmaceutical market; a high level of competition, as a result of which there is a shift in competitiveness factors from the level of the product to the level of the organization as a whole; an increase in the number of interactions in the process of drug circulation; social responsibility of business and its customer orientation; the characteristics of pharmaceutical products, as well as the requirements of regulatory legal acts, are the main reasons for the development and implementation of quality assurance systems or, at a higher level of development, quality management in the implementation of pharmaceutical activities.
An essential condition for the functioning of the sphere of drug circulation, as well as one of the main mechanisms for ensuring the required level of quality and safety of pharmaceutical products and services in the interests of the consumer, is the standardization procedure [1-2].
Standardization is the activity of establishing rules and characteristics for the purpose of their voluntary reuse, aimed at achieving orderliness in the areas of production and circulation of products and increasing competitiveness.
To date, one of the pressing issues of healthcare in the Republic of Uzbekistan is the provision of vital drugs that meet the high requirements of modern medicine. The quality of infusion solutions must meet the stringent requirements of modern standards. Only in the conditions of pharmaceutical production, it is possible to eliminate the influence of the human factor as much as possible and introduce multi-stage quality control. The control of the main indicators of the quality of the finished product and the parameters of the technological process plays an important role in obtaining drugs of guaranteed quality. An increased risk in the parenteral route of administration of large volume solutions (100 ml or more) or infusion solutions causes high requirements for their quality [3].
Quality standards for medicines should ensure the development of a high quality, effective and safe medicine, and should be revised in a timely manner, taking into account new achievements in medical, pharmaceutical and other sciences and the requirements of leading foreign pharmacopoeias.
An objective assessment of the quality of medicines depends not only on the merit of the methods, but also on the fact that their use in different laboratories allows obtaining identical results. This is ensured by the standardization of quality assessment methods, the preparation of solutions, reagents and indicators, the standardization of instruments, etc. The standard as ND establishes a set of norms or requirements for the object of standardization [4].
Braecord is a combined drug containing in its composition: sodium citicoline, L-arginine hydrochloride, levocarnitine, used as a nootropic and metabolic agent.
Purpose of the study. The purpose of these studies is to develop methods for assessing the quality and establishing indicators of the quality of the combined drug "Bralecord" solution for infusion.
## II. EXPERIMENTAL PART
### a) Materials and research methods
As objects of study, 5 series of pilot samples of the drug "Bralecord" solution for infusions were used. During the study, solvents, reagents and consumables from MERCK (Germany) were used. The following auxiliary equipment was also used in the tests: magnetic stirrers, BP-310S electronic analytical balance from Sartorius (Germany), HS 32 AC sterilizer with automation, Seven Easy pH meters from Mettler Toledo (Switzerland) and 766 Calimatic Knick (Germany), Julabo water thermostat (Germany), Osmomat 010 type osmometer, Gonotek (Germany), PAMAS SVSS liquid particle counters (Germany).
### b) Results and its discussion
The evaluation of the quality indicators of the study drug was carried out in accordance with the modern requirements of the national and foreign pharmacopoeias [5-7], as well as in accordance with the general technical regulation on the safety of medicines [8] in terms of such indicators as:
description, authenticity, transparency, color, pH, mechanical inclusions, impurities, osmolality, sterility, bacterial endotoxins, abnormal toxicity, quantitation.
The initial stage of work is organoleptic control, which is a mandatory type of control and consists in checking the medicinal product in terms of appearance, smell, mixing uniformity, and the absence of mechanical impurities in liquid dosage forms.
### c) Description
The drug should be a clear, colorless or slightly yellowish solution (visual method). All studied five series of the drug meet the requirements of the pharmacopoeia.
### d) Authenticity
Identification was carried out by a chemical method using qualitative reactions to the main components of the drug: sodium citicoline, arginine, levocarnitine and chlorides.
Sodium citicoline was determined by the spectrophotometric method at an absorption maximum at $280~\mathrm{nm}$. $1.0~\mathrm{ml}$ of the drug was placed in a volumetric flask with a capacity of $100~\mathrm{ml}$, the volume of the solution was brought to the mark with a $0.01~\mathrm{mol} / \mathrm{l}$ solution of hydrochloric acid and mixed. $10~\mathrm{ml}$ of the resulting solution was placed in a volumetric flask with a capacity of $50~\mathrm{ml}$, the volume of the solution was brought to the mark with a $0.01~\mathrm{mol} / \mathrm{l}$ solution of hydrochloric acid and stirred (test solution).
The optical density of the resulting solution was measured on a spectrophotometer at the absorption maximum at a wavelength of $280\mathrm{nm}$ in a cuvette with a layer thickness of $10\mathrm{mm}$, using a $0.01\mathrm{mol}/$ hydrochloric acid solution as a reference solution. In parallel, the optical density of the RSO solution of sodium citicoline was measured at the same wavelength.
Arginine. 15 ml of water was added to 5 ml of the preparation. To 2 ml of the resulting solution was acidified 1 ml of α-naphthol solution and 2 ml of a mixture of equal volumes of 3% sodium hypochlorite solution and water; a red color is formed.
Levocarnitine. 2 ml of the drug was transferred into a test tube, 5 ml of 1 mol/l hydrochloric acid solution and a few drops of ammonium rheincate solution were added, and a red-violet precipitate formed.
Preparation of ammonium rheincate solution: about $500\mathrm{mg}$ of ammonium rheincate was mixed with $20\mathrm{ml}$ of water, shaken periodically for 1 hour and filtered. The solution is used within 2 days.
Chlorides. $2.0 \, \text{ml}$ of the drug was acidified with dilute nitric acid, $0.4 \, \text{ml}$ of a silver nitrate solution was added, and a white curled precipitate (chlorides) was formed upon standing.
All studied five batches of the drug confirmed the presence of citicoline sodium, arginine, levocarnitine and chlorides in the solution of the study drug.
Transparency. The solution of the drug should be transparent compared to water for injection.
Chromaticity was determined in accordance with the European or British Pharmacopoeia. It was found that the color of the preparation should not be more intense than the reference solution Y7.
The pH was set potentiometrically. It was found that the pH of the drug solution should be in the range from 5.0 to 7.5.
All studied five batches of the drug met the requirements of pharmacopoeias in terms of mandatory indicators: transparency, color, pH.
#### Foreign impurities
Citicoline sodium. High performance liquid chromatography method.
Chromatographic system and determination conditions Instrument liquid chromatograph "ShimadzuLC-6A"
Steel column, size $30~\mathrm{cm} \times 4.0~\mathrm{mm}$
Octadecylsilane filler chemically bonded to porous silica gel or ceramic microparticles $10\mu \mathrm{m}$ in diameter (LI, USP)
Flow rate 2 ml/min
Detection 275 nm
Injection volume $20\mu l$
A column filled with octadecylsilane silica gel and phosphate buffer solution was used (equal volumes of $0.1 \, \text{mol/l}$ potassium dihydrogen phosphate and tetrabutylammonium phosphate solution were mixed, the pH value of $0.01 \, \text{mol/l}$ tetrabutylammonium hydroxide was adjusted to 4.5 with phosphoric acid), methanol $(95:5)$ as mobile phase.
1 ml of the drug was carefully transferred into a 10 ml volumetric flask and diluted with water to the mark (test solution).
Dissolve the required amount of citicoline sodium RSO and levocarnitine RSO in water to obtain a solution of $1.045 \, \mathrm{mg/ml}$ sodium citicoline and $2.0 \, \mathrm{mg/ml}$ levocarnitine (control solution).
Transfer $1.25 \mathrm{ml}$ of the control solution to a 100 ml volumetric flask. Dilute to the mark with water and mix well (system suitability solution).
Introduced $20\mu l$ of reference solution 1 into the column. The dilutions were adjusted so that the main maximum in the chromatogram was $20 - 25\%$ of the full scale of the chart. Add $20\mu l$ of each test solution, system suitability solution, and control solution separately to the column. Chromatography was continued at 2.5 times the retention time of the citicoline peak.
#### Calculations:
The content of an individual non-identifying or identifying impurity, in percent:
$$
X = \frac{AT\times 100\%}{AS}
$$
where: $\mathrm{AT} =$ Peak area of an individual impurity in the test solution As = Sum of all peak areas in control solution
At the same time, it was established that the individual impurity should be no more than $0.5\%$, and the total impurity - no more than $2.0\%$. All five batches of the study drug met the established norm.
Substances detected by ninhydrin. The determinations are carried out by thin layer chromatography.
System suitability solution: $0.4\mathrm{mg / ml}$ each of arginine hydrochloride and L-lysine hydrochloride with water
Sample solution: 10 mg/ml RSO arginine hydrochloride with water
Standard solution: dilute 1 ml of sample solution with water to 100 ml. Dilute 5 ml of the resulting solution with water to 10 ml (0.05 mg/ml)
Note: The concentration of the solution is approximately $0.5\%$ of this sample solution.
Test solution: $100 \mu l$ of the drug is added to $320 \mu l$ of water $(10 \mathrm{mg} / \mathrm{ml})$
Chromatography System Mode: TLC
Adsorbent: 0.25 mm layer of chromatographic silica gel mixture
Applied sample volume: $5\mu l$
Mobile solvent system: isopropyl alcohol and ammonium hydroxide (70:30)
Nebulizer: 2 mg/ml ninhydrin in butyl alcohol and 2N acetic acid (95:5)
Drying: at a temperature from $100^{\circ}\mathrm{C}$ to $105^{\circ}\mathrm{C}$ until complete removal of ammonia. The plate is sprayed with a solution of $2\mathrm{mg/ml}$ ninhydrin in a mixture of butyl alcohol and $2\mathrm{N}$ acetic acid (95:5) and heated at $100^{\circ}\mathrm{C}$ to $105^{\circ}\mathrm{C}$ for 15 minutes. Examine the plate under daylight. The system suitability chromatogram shows two completely separated spots.
Norm: any spot, except for the main one, should not be more intense than the main spot on the chromatogram of the standard solution (0.5%). Individual impurities: no more than 0.5%, total impurities: no more than 2.0%.
The results of the analysis are considered reliable if two clearly separated spots appear on the chromatogram of the system suitability solution.
All studied five series of the drug corresponded to the established norm.
Levocarnitine. High performance liquid chromatography method.
Reagents: 2M sodium hydroxide solution. Solution A: $6.81 \mathrm{~g}$ of $\mathrm{KH}_{2} \mathrm{PO}_{4}$ are dissolved in $800 \mathrm{ml}$ of water, 2M sodium hydroxide solution is added to obtain a pH of
4.7, the volume of the solution is adjusted to the mark with water and mixed; Acetonitrile.
Chromatography conditions. Column: Aminopropyl methylsilangel (USP L8), 250x4.6mm, $5\mu \mathrm{m}$; Column temperature: $30^{\circ} \mathrm{C} + 1^{\circ} \mathrm{C}$; UV detector: 205nm. Mobile phase: A mixture of 35 volumes of solution A and 65 volumes of acetonitrile; flow rate: 1.0ml/min; injection volume: $25\mu \mathrm{l}$.
Standard solutions and test solution are introduced into the chromatograph and chromatograms are recorded within 20 minutes. The sensitivity of the system is determined by the height of the main peak on the chromatogram of the standard solution (c), which is at least $20\%$ of the full scale of the recording device. The test is considered invalid if the resolution between the peaks of levocarnitine and levocarnitine impurity A in the chromatogram of the standard solution (c) is 0.9. On the chromatogram of the test solution: the area of any peak of levocarnitine impurity A is not more than the main peak of the chromatogram of the standard solution (c) $(1\%)$; the area of any peak, except for the main peak and the peak of levocarnitine impurity A, does not exceed the area of the main peak in the chromatogram of the standard solution a $(0.2\%)$; the sum of the areas of all peaks, except for the main peak and the peak of impurity A, does not exceed 2.5 times the area of the main peak in the chromatogram of standard solution a $(0.5\%)$.
Preparation of standard solution "a". 100 mg of the drug solution is transferred into a 100 ml volumetric flask, the volume is adjusted to the mark with solution A. 1 ml of the resulting solution is diluted to 10 ml with the same solvent (0.2%).
Preparation of standard solution "c". 20.0 mg of the working standard sample of levocarnitine impurity A is dissolved in water, the solution is adjusted with water to 100 ml. 2.5 ml of the resulting solution is diluted to 10 ml with solution A (1.0%).
Preparation of standard solution "c". 10 mg of the working standard sample of levocarnitine impurity A is dissolved in water and diluted with water to 25 ml and 2.5 ml of the resulting solution is diluted to 10 ml with solution A.
Preparation of standard solution "d". Dissolve $100\mathrm{mg}$ of the working standard of levocarnitine in $10\mathrm{ml}$ of standard solution C.
Preparation of the test solution. 2.5 ml of the preparation solution is transferred into a volumetric flask with a capacity of 100 ml and the volume is brought to the mark with solution A.
Norm: impurities A should be no more than $1.0\%$; other impurities - no more than $0.2\%$; the sum of impurities, except for impurity A - no more than $0.5\%$.
According to the results of the studies, all five series of the drug under study corresponded to the established norm.
Osmolality. The determination of the osmolality of the solution is carried out by the cryoscopic method using a Beckmann thermometer. The determination of the freezing point is carried out on the installation shown in Figure 1.
The main part of the setup is a test tube with a side branch. Its upper opening is tightly closed with a cork through which the Beckmann thermometer and a wire stirrer pass, one end of which is bent in the form of a ring freely covering the lower part of the thermometer. This test tube is inserted into a wider test tube, which acts as an air jacket that prevents the liquid from cooling too quickly. The assembled apparatus is placed in a Bunsen beaker, which is filled with a cooling mixture before the experiment.
The role of the cooling mixture is performed by ice chips, to which crystalline sodium chloride is added to reduce the temperature. A stirrer is used to stir the cooling mixture. The temperature of the cooling mixture should be $(4 - 5)^{\circ}C$ below the freezing point of the test liquid.
The zero point of the instrument is set to water for injection. The instrument is calibrated using standard sodium chloride solutions. The determination is carried out three times and the average value is taken.
As needed, prepare standard solutions based on the data in Table 1.
To determine the freezing point of a solvent or a test solution, $28 - 30\mathrm{g}$ of liquid is placed into an inner small test tube through a side hole, a stirrer and a Beckmann thermometer are also placed here. This tube is placed through an air jacket into the cooled mixture and the test liquid is evenly mixed by raising and lowering the stirrer. When determining the freezing point of a liquid, the mercury column in the thermometer begins to fall as the liquid cools. Usually, before freezing, the liquid is supercooled, and the temperature of the liquid drops below the freezing point. As soon as the crystallization process begins, the temperature of the solution rises beyond the freezing point.
Table 1: Data for osmometer instrument
<table><tr><td>Mass of sodium chloride, in grams per 1 kg of water</td><td>Actual osmolarity (mosm/kg)</td><td>Theoretical osmolality (ideal)</td><td>Molar osmotic coefficient (mosm/kg)</td><td>Freezing temperature drop ΔTzam.</td></tr><tr><td>3,087</td><td>100</td><td>105,67</td><td>0,9463</td><td>0,186</td></tr><tr><td>6,260</td><td>200</td><td>214,20</td><td>0,9337</td><td>0,372</td></tr><tr><td>9,463</td><td>300</td><td>323,83</td><td>0,9264</td><td>0,558</td></tr><tr><td>12,684</td><td>400</td><td>437,07</td><td>0,9215</td><td>0,744</td></tr><tr><td>15,916</td><td>500</td><td>544,66</td><td>0,9180</td><td>0,930</td></tr><tr><td>19,147</td><td>600</td><td>655,24</td><td>0,9157</td><td>1,116</td></tr><tr><td>22,380</td><td>700</td><td>765,86</td><td>0,9140</td><td>1,302</td></tr></table>
The increase in temperature occurs due to the release of open heat of solidification.
After that, the test tube is removed from the liquid, the crystals are melted by heating the test tube with the hand, and the determination is repeated again.
The experiment is carried out three times. The discrepancy between the definitions should be no more than $0.01^{\circ}\mathrm{C}$. In case of hypothermia, it is necessary to add a solvent crystal to the liquid. The accuracy of determination with this method is $+5\%$.
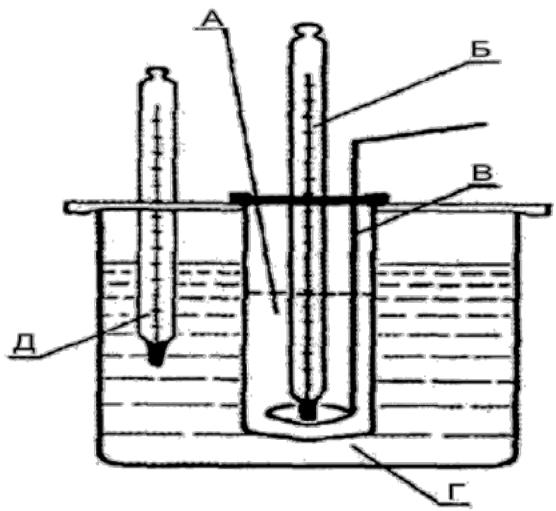
The device of the Beckman device: A - a vessel for the test solution; B - Beckman thermometer; B - stirrer; G - container with coolant; D - thermometer for measuring the temperature of the cooling mixture.
Before each measurement, the tube with the lateral process is rinsed with the solution intended for the study, and the measurement is also made with the test samples.
The instrument is ready to measure the test product if the value obtained for the calibration solutions is within two values of the calibration scale. To reduce the error and check the reproducibility, it is recommended to repeat measurements with several samples from the same sample, averaging the results. The measurement error should not exceed $\pm 2\%$.
The calculation of osmolality is carried out according to the following formula:
$$
\mathrm {S} _ {\mathrm {o s m}}, \mathrm {m O s m o l / k g (m O s m o l / l)} = \frac {(\mathrm {T 2 - T 1}) \times 1 0 0 0}{1 . 8 5 8},
$$
where: 1.858 is the molar cryoscopic constant of water, corresponding to the decrease in freezing point that occurs as a result of dissolution
1 mole of a substance in $1\mathrm{kg}$ of water;
1000 - Conversion factor osm/kg to mosm/kg;
$\mathrm{T}_{2}$ - freezing point of pure solvent (OC);
$T_{1}$ is the freezing point of the solution (0C).
For 200 ml vial:
<table><tr><td>Particlesize</td><td>Number of particles in 1 ml</td></tr><tr><td>10 micronsormore</td><td>Not more than 25 h/ml</td></tr><tr><td>25 micronsormore</td><td>Not more than 3 h/ml</td></tr></table>
All studied five series of the drug corresponded to the established norm in terms of "mechanical inclusions".
Based on the measurements carried out, it was found that the osmolality should be in the range from $410 \, \text{mOsmol/kg}$ to $590 \, \text{mOsmol/kg}$. All studied five series of the drug corresponded to the established norm.
Mechanical inclusions. Mechanical inclusions in dosage forms for parenteral use are extraneous mobile insoluble particles, with the exception of gas bubbles, accidentally present in drug solutions. The test for the presence of visible and invisible particulate matter is intended for visual evaluation of liquid parenteral dosage forms, including infusion solutions, and is a mandatory pharmacopoeial quality indicator.
Based on the foregoing, as a result of the research, visible and invisible mechanical particles were determined. To detect visible particles in accordance with the SP RUz., Eur.F., 2.9.20, OFS 42 Uz-0006-3341-2018, during visual inspection, the test preparation should not contain visible particles (visible particles should be absent).
To detect invisible particles in accordance with the SP RUz., Eur.F., 2.9.19, OFS 42 Uz-0005-3340-2018, invisible particles per 50 ml and 100 ml bottle for particles $\geq$ 25 microns should be no more than 600 pieces, for particles $\geq$ 10 microns should be no more than 6000 pieces (Ev.F., 2.9.19, OFS 42 Uz-0005-3340-2018).
The results of studies on the quality indicators of the drug "Bralecord" solution for infusion are presented in table 2.
Table 2: Research results on some quality indicators drug "Bralecord" solution for infusion
<table><tr><td>The name of indicators</td><td>Norms</td><td>Series 1</td><td>Series 2</td><td>Series 3</td><td>Series 4</td><td>Series 5</td></tr><tr><td>Description</td><td>Clear, colorless or slightly yellowish solution.</td><td>Corresponds</td><td>corresponds</td><td>corresponds</td><td>corresponds</td><td>corresponds</td></tr><tr><td>Authenticity</td><td>SF-metry, qualitative reactions</td><td>confirmed</td><td>confirmed</td><td>confirmed</td><td>confirmed</td><td>confirmed</td></tr><tr><td>Transparency</td><td>The drug must be transparent</td><td>transparent</td><td>transparent</td><td>transparent</td><td>transparent</td><td>transparent</td></tr><tr><td>Chroma</td><td>The color of the preparation should not be more intense than the reference solution Y7</td><td>no more intense than reference solution Y7</td><td>no more intense than reference solution Y7</td><td>no more intense than reference solution Y7</td><td>no more intense than reference solution Y7</td><td>no more intense than reference solution Y7</td></tr><tr><td>pH</td><td>5,0-7,5</td><td>6,05</td><td>6,10</td><td>6,08</td><td>6,12</td><td>6,02</td></tr><tr><td colspan="7">Foreign matter:</td></tr><tr><td>Citicoline sodium:</td><td>no more than 0,5%</td><td>no more than 0,5%</td><td>no more than 0,5%</td><td>no more than 0,5%</td><td>no more than 0,5%</td><td>no more than 0,5%</td></tr><tr><td>Individual impurity total impurity</td><td>no more than 2,0%</td><td>no more than 2,0%</td><td>no more than 2,0%</td><td>no more than 2,0%</td><td>no more than 2,0%</td><td>no more than 2,0%</td></tr><tr><td>Levocarnitine admixture A: others: the sum of impurities, except for impurity A:</td><td>no more than 1,0% no more than 0,2% no more than0,5%</td><td>no more than 1,0% no more than 0,2% no more than0,5%</td><td>no more than 1,0% no more than 0,2% no more than0,5%</td><td>no more than 1,0% no more than 0,2% no more than0,5%</td><td>no more than 1,0% no more than 0,2% no more than0,5%</td><td>no more then 1,0% no more than 0,2% no more than0,5%</td></tr><tr><td>Substances detectedbyninhy drin</td><td>Any spot, except for the main one, should not be more intense than the spot on the chromatogram of the standard solution (0.5%)</td><td>no more than0,5%</td><td>no more than0,5%</td><td>no more than0,5%</td><td>no more than0,5%</td><td>no more than0,5%</td></tr><tr><td>Osmolality</td><td>410-590 mosmol/l</td><td>475mosmol/l</td><td>478 mosmol/l</td><td>476 mosmol/l</td><td>477 mosmol/l</td><td>480 mosmol/l</td></tr><tr><td>Mechanicalinc lusions (visible)</td><td>Visible particles must be absent</td><td>missing</td><td>missing</td><td>missing</td><td>missing</td><td>missing</td></tr><tr><td>Mechanicalinc lusions (invisible)</td><td>One vial for particles ≥ 25 microns should contain no more than 600 pieces; Particles ≥ 10 microns should be no more than 6000 pieces</td><td>corresponds</td><td>corresponds</td><td>corresponds</td><td>corresponds</td><td>corresponds</td></tr></table>
Foreign matter: Citicoline sodium: Individual total impurity Levocarnitine admixture A: others: the sum of impurities, except for impurity A: Substances detected by inhy drin Osmolality Mechanical inc lusions (visible) Mechanical inc lusions (invisible)
## III. CONCLUSION
On the basis of the conducted experimental studies, the main indicators of the quality of the drug "Bralecord" solution for infusions were established in terms of: description, authenticity, transparency, color, pH, impurities, osmolality, mechanical inclusions. The limits of their normalization in accordance with the pharmacopoeias have been established. Approved quality indicators are included in the regulatory documentation for the study drug.
Generating HTML Viewer...
References
8 Cites in Article
Victoria Muravyeva,Natalya Soboleva,Artem Zavyalov,Oleg Makhlis,Tatyana Likhikh,Irina Kis (2008). Comparative characteristics of quantitative methods for the determination of flavophospholipol in medicinal preparations by the agar diffusion method.
N Tyukavkina (2008). Standardization and quality control of medicines.
E Moiseeva (2004). Fig. 4. Abstract of the Dissertation for the Degree of Candidate of Medical Sciences by Yu.D. Zilber.
E Krasnov,T Kadyrov (2005). Standardization of medicines.
(2021). Unknown Title.
American Pharmacopeia.
(2016). British Pharmacopoeia and the European Pharmacopoeia.
Herolind Jusufi,Nicholas Boivin (2023). Navigating Access and Optimizing Medication Infusions in an Academic Medical Center: A Quality Improvement Study.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Surayyo Z. Yuldasheva. 2026. \u201cEvaluation of the Quality Indicators of the Drug “Bralekord” Solution for Infusions\u201d. Global Journal of Medical Research - B: Pharma, Drug Discovery, Toxicology & Medicine GJMR-B Volume 23 (GJMR Volume 23 Issue B1).
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.