This study aimed to determine whether the hepatitis C virus (HCV) infects human megakaryocytes and platelets and to measure the expression of receptors involved in virus-cell interaction. Platelets from healthy donors were infected with HCV in vitro and analyzed for viral expression and the receptors claudin-1 and cluster of differentiation 81 (CD81). HCV was detected on the surface and cytoplasm of both cells; cytoplasmic expression was higher compared to the surface. Platelets presented a claudin-1+/CD81-phenotype, and megakaryocytes showed aclaudin-1+/CD81+ phenotype. We conclude that megakaryocytes and platelets are susceptible to HCV infection, regardless of CD81 expression, and megakaryocytes may serve as possible sites of viral replication.
## I. INTRODUCTION
Despite significant advances in the treatment of chronic hepatitis C with direct-acting antiviral therapy [1,2], approximately 290,000 people died from disease complications in 2019, mainly from causes related to cirrhosis and hepatocellular carcinoma. Around 58 million people chronically carry the hepatitis C virus (HCV), and annually it is reported 1.5 million new cases. Therefore, hepatitis C is still a global public health problem [3].
HCV is hepatotropic and mainly targets hepatocyte cells. However, extrahepatic viral locations are reported in endothelial cells [4], gastrointestinal mucosal cells [5], fibroblasts [6], macrophages, dendritic cells [7-11], erythrocytes [12], peripheral blood mononuclear cells (especially B- and T-lymphocytes, monocytes [13]), platelets [13;14] and megakaryocytes [2]. The existence of extrahepatic reservoirs may help to understand the symptoms or extrahepatic manifestations that patients develop. Moreover, extrahepatic reservoirs may be related to the high rate of chronic infection [2,4,14-17]. Thrombocytopenia is a common extrahepatic manifestation among patients with chronic hepatitis C, particularly those with long-term complications. This condition increases the risk of bleeding and complicates liver biopsies, which leads to compromised patient follow-up [18,19]. Acquired thrombocytopenia is triggered by several causes, including diseases caused by infectious agents such as HCV, HIV, and Helicobacter pylori [20,21]. The pathophysiology of thrombocytopenia may involve factors such as increased platelet destruction, decreased platelet production, bone marrow suppression, hypersplenism (including platelet sequestration in the enlarged spleen secondary to portal hypertension), autoimmunogenicity, thrombopoietin production imbalance, and adverse therapeutic effects [18, 22].
HCV's presence in bone marrow [22] and platelets [23] suggests these areas as biological compartments for the virus. Thrombocytopenia in chronic hepatitis C patients is associated with HCV's infection of, or adhesion to, platelets [14,23]. This interaction impairs both platelet function and the production of megakaryocytes, which are essential for platelet formation [2,25]. Platelet recovery after viral suppression/eradication further indicates the involvement of virus-platelet interactions in thrombocytopenia [20]. Patients with chronic hepatitis C also present alterations in the bone marrow microenvironment. Patients with a high viral load present hypo- and hypercellularity associated with sedimentation of immune complexes and peripheral cytopenia [22]. However, the exact mechanism of the HCV interaction with platelets—whether the virus only adsorbs to the platelet membrane or is internalized there—is poorly understood. Additionally, the virus can infect megakaryocytes in the bone marrow, transferring to platelets during thrombocytogenesis. Evidence of HCV in bone marrow [22] and platelets [23], along with susceptibility to in vitro infection in the megakaryocytic lineage (MEG-01) [2] and platelets [14] supports this view.
Considering the lack of evidence regarding the intra- and extracellular HCV localization and the receptors involved in the virus-target cell interaction, we evaluated the presence of intracellular and extracellular HCV in megakaryocytes and platelets, and the expression of the receptors used by the virus, cluster of differentiation 81 (CD81) and claudin-1. We sought to clarify whether HCV is only adsorbed on the surface of megakaryocytes and platelets or whether these cells are susceptible to infection. Moreover, we explored the potential link between this viral interaction and the thrombocytopenia often observed in infected patients.
## II. MATERIAL AND METHODS
### a) Ethical aspects
The Research Ethics Committee of the Botucatu Medical School (UNESP) approved this study under protocol 1.354.285. All donors and patients participating in the study provided their consent by signing informed consent forms. All experimental assays were performed at the Laboratory of Applied Biotechnology, Clinical Hospital of the Botucatu Medical School, UNESP.
### b) Megakaryocyte and platelet isolation
Megakaryocytes were sourced from six bone marrow donors at Amaral Carvalho Hospital in Jaú, Brazil. These cells were harvested from the donors' iliac crest concurrently with their bone marrow donation for transplantation. Bone marrow samples (4 mL) were collected with modified CPDA-1 anticoagulant (CPDA-1 with 6% EDTA) diluted 1:1 with megakaryocyte buffer (PBS solution with 1% D-glycoside, 3% sodium citrate and 13.5% bovine serum albumin at 22%) [26,27,28]. The diluted bone marrow was sterile filtered through a nylon filter (160μm) and carefully placed on Percoll® (Sigma-Aldrich) in equivalent proportions (1:1). The samples were then centrifuged at 405xg for 20 minutes at 20°C. Megakaryocyte pellets were collected from the top layer of Percoll® and immediately washed with the double volume of megakaryocytes buffer (4°C). All washed pellets were centrifuged at 405xg for 10 minutes at 4°C. The enriched megakaryocyte pellet was resuspended in Roswell Park Memorial Institute Medium (RPMI) 1640 with L-glutamine and 1% antibiotics (Gibco) until in vitro infection.
Peripheral blood platelets were collected with EDTA anticoagulant (BD Vacutainer®- 5mL) from healthy donors $(n = 4)$ and processed as described by Padovani et al.[14]. Briefly, fresh blood samples were centrifuged at $700\mathrm{Xg}$ for 3 minutes at room temperature to obtain platelet-rich plasma, followed by a second centrifugation at $1600\mathrm{Xg}$ for 5 minutes (room temperature) to obtain platelet pellet. Cells were washed four times with saline $(0.9\%$ NaCl) and resuspended in RPMI 1640 supplemented with L-glutamine and $1\%$ antibiotics (Gibco).
### c) In vitro infection of megakaryocytes and platelets
For in vitro megakaryocyte infection, $4 \times 10^{6}$ nucleated cells of the enriched megakaryocyte pellet were resuspended in $1 \mathrm{ml}$ of RPMI 1640 medium. Then $6 \mathrm{mL}$ of HCV plasma genotype 1 (100,000 viral RNA copies/mL) was added to the flask culture. Cells were incubated for 36 hours in a conventional cell incubator (Thermo Fisher) at $37^{\circ} \mathrm{C}$ in $5\% \mathrm{CO}_{2}$. During the infection period, the cell culture was or bitally agitated (18 hours) without changing the culture medium. HCV-negative plasma was used as a control.
In vitro platelet infection was performed as described by Padovani et al. [14]. Briefly, $1\mathrm{mL}$ of resuspended platelets was incubated with $1\mathrm{mL}$ of genotype 1 HCV plasma containing 100,000 RNA copies/mL from patients with positive RT-PCR. Samples were incubated in a shaker (New Brunswick Scientific) for 48 hours at $37^{\circ}\mathrm{C}$ with continuous shaking at $10\mathrm{xg}$.
### d) Assessment of HCV infectivity in megakaryocytes and platelets
The presence of HCV in megakaryocytes and platelets was examined following in vitro infection. This analysis was conducted using flow cytometry and confocal microscopy. The purpose of these assessments was to confirm the susceptibility of these cells to the virus and to determine the viral location. For HCV detection, we used the monoclonal antibody NS4A-FITC (clone S4-13 - Abcam), a protein common to all HCV genotypes [29]. Molecular biological assay was performed only in megakaryocytes, since previous study has shown HCV RNA expression in platelets, as described in Padovani et al. [14].
## i. Flow cytometry analysis
Following infection, megakaryocyte cells were labeled with anti-human-CD61-PE (VIPL2 clone, EXBIO) and anti-CD45-PerCP (HI30 clone, BD Pharmingen) [26]. To label platelets, we used only the anti-human-CD61-PE (VIPL2 clone, EXBIO), a commonly used marker for both platelets and megakaryocyte cells. The presence of HCV was evaluated using the monoclonal anti-hepatitis C virus antibody NS4A-FITC (S4-13 clone -Abcam). Cells were incubated for 30 minutes at room temperature in the dark, according to the manufacturer's instructions. In this assay, the control group comprises megakaryocyte cells incubated with plasma without HCV. Peripheral platelets obtained from HCV patients $(n = 2)$ were used to compare the virus behavior in vitro and in vivo. To detect HCV, we first labeled the surface of megakaryocyte and platelet cells. Then, we fixed the cells with $4.2\%$ paraformaldehyde for 30 minutes at room temperature. Both cell types were permeabilized with $0.4\%$ Triton X-100 for 5 minutes at room temperature and then stained with anti-NS4A. During the steps, we washed the cells with the respective buffers following the rotation described above. To prevent the formation of platelet clots, polystyrene tubes were coated with $22\%$ bovine albumin [30]. We obtained both cell types using a FACSCaliburTM device (BD Bioscience) and analyzed the results using CellQuestTM and FlowJoTM software (BD Bioscience). 50,000 events were collected at the CD61+ gate for all samples. The isotype controls were conducted following the experimental protocol and included Mouse IgG1-FITC (clone MOPC-21), Mouse IgG1-PerCP (clone MOPC-21), and Mouse IgG1-APC (clone MOPC-21 - BD Pharmingen).
The intra- and extracellular expression of HCV antigen (NS4A) was determined by comparing the mean fluorescence intensity (MFI - absolute number) values using the flow cytometry crossmatch assay model[28] $\frac{MIF NS4A intracellular}{MIF NS4A surface}$. Expression index value $>1.0$ indicates intracellular expression due to the increase in fluorescence compared to surface expression.
## ii. Indirect immunofluorescence staining of HCV-infected cells (confocal microscopy)
Twenty-five microliters of labeled megakaryocytes and platelets were applied to silanized HDA slides and covered with Fluoroshield histology mounting medium (F6182-Sigma-Aldrich). The slides were sealed with resin, stored at $6^{\circ}\mathrm{C} \pm 2^{\circ}\mathrm{C}$, and protected from light. Paraffin-embedded liver fragments from HCV-positive patients' biopsies were used as a positive control in this experiment. First, paraffin-embedded liver tissues were sectioned at a thickness of $0.3\mu \mathrm{m}$ and fixed onto silanized HDA slides. The slides were then incubated using an antigen retrieval PT Link device (Dako) for 60 minutes at $65^{\circ}\mathrm{C}$ in a histological incubator at the Immunohistochemistry Laboratory of the Department of Pathology of the Clinical Hospital of the Botucatu Medical School - UNESP. To block nonspecific binding, avidin and biotin (Vector Laboratories) were applied to coverslip slides for 20 minutes at room temperature. The slides were washed with PBS and labeled for two hours at room temperature with antihuman CD61-PE diluted 1:50 with EnVisionTM FLEX Antibody Diluent (Dako). After labeling, slides were washed and incubated overnight with EnVisionTM diluted monoclonal anti-hepatitis C virus antibody NS4A-FITCDiluted 1:20. Then, the slides were covered with Fluoroshield Histology Mounting Medium (Sigma-Aldrich) and kept at $4^{\circ}\mathrm{C}$ until confocal analysis. Images were taken using the TCS SP5 - Leica Laser Scanning Confocal Microscope, using the LAS AF software version 2.7.3.9723, available at the Electronic Microscopy Center of the Botucatu Institute of Biosciences, UNESP.
iii. Detection of HCV RNA in megakaryocytes using real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR)
Part of the megakaryocyte-enriched cell suspension was divided equally. Tube 1 (negative control) was incubated with $1\mathrm{mL}$ of HCV-negative plasma. Tube 2 (test condition) had megakaryocyte added to RPMI and incubated with a pool of plasma containing $100,000~\mathrm{IU / mL}$ of HCV genotype 1. Tube 3 (negative control) had $1\mathrm{mL}$ of the $100,000~\mathrm{IU / mL}$ HCV genotype 1 plasma pool. The purpose of the final control assay was to prevent the adsorption of RNA present in the plasma in the tube; therefore, no megakaryocyte cells were added. The tubes were washed five times to remove any free virus that did not aggregate in the cells. All supernatants from each step were collected and frozen at $-80^{\circ}\mathrm{C}$ for subsequent quantification of HCV RNA by qRT-PCR using the Abbott RealTime HCV assay (Abbott Molecular). The genomic region of the HCV $5^{\prime}$ UTR was analyzed by nested PCR reaction, with HCV RNA being converted to complementary DNA using the High-Capacity cDNA Archive kit (Applied Biosystems). All procedures were carried out according to the instructions provided by the manufacturer.
### e) Evaluation of the expression of key HCV entry receptors claudin-1 and CD81
We evaluated the expression of Claudin-1 and CD81, key cellular entry receptors used by HCV to infect cells. Flow cytometry and confocal microscopy were used to examine megakaryocytes and platelets receptors. To perform immunophenotyping, monoclonal antibodies were used along with phenotypic markers, as previously described. Specifically, anti-human Claudin1Alexa Fluor 488 (clone 2H10D10, RheaBiotech) and anti-human CD81-APC (clone M38, EXBIO) were used.
### f) Statistical analysis
Descriptive statistical analysis, including means, standard deviations, and ranges (minimum and maximum), was performed with Prism 8 software (GraphPad®).
## III. RESULTS
### a) HCV-infected megakaryocytes and platelets
Efficient HCV infection of megakaryocytes and platelets was demonstrated in vitro through the detection of viral expression (NS4A) and viral load (mean 34.25 IU/mL) [31]. The objective was to demonstrate the biological event through descriptive analysis. In megakaryocytes, the average percentage of NS4A in the membrane was $51.71\% \pm 26.72$ (range $11.01\% - 85.96\%$ ). For the cytoplasm, this percentage was $75.57\% \pm 29.66$ (range $26.61\% - 99.77\%$ ) (Figure 1-A). The NS4A MFI in the membrane was $30.23 \pm 9.03$
(range 19.11-43.71), while in the cytoplasm, the MFI was $63.32 \pm 32.41$ (range 21.29-107.5).
The expression of intracellular viral NS4A was 2.09 times higher than that of membrane NS4A (Figure 1-B), indicating a higher concentration of NS4A or HCV in the intracellular compartment. Supporting this observation, confocal microscopy images showed the presence of NS4A on the megakaryocyte membrane
(Figure 1-C1), along with CD61 (a megakaryocyte characterization marker) (Figure 1-C2) and 7AAD (a nuclear marker) (Figure 1-C3). By examining the Z-axis (depth) of the cells, we observed a high brightness density of NS4A expression inside the cell (Figure 1-C4), corroborating the findings from the flow cytometer analysis.
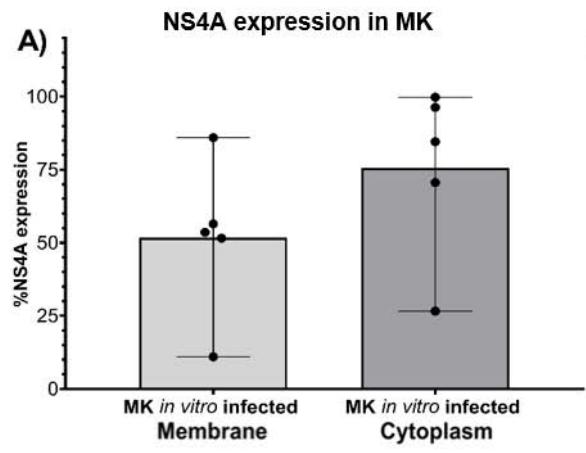
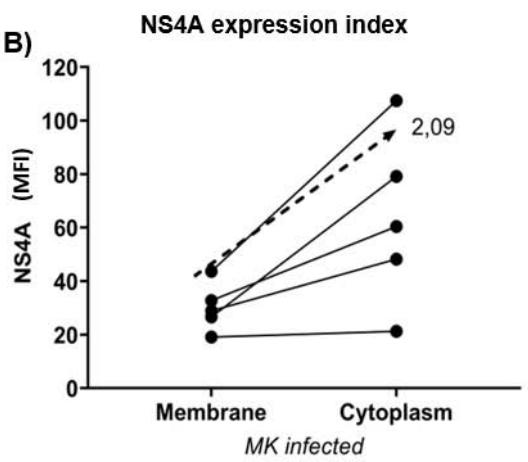
 C) Figure 1: In vitro HCV-infected megakaryocytes were examined by flow cytometry and confocal microscopy. The percentage of NS4A expression was $51.71\% \pm 26.72$ (range $11.01\% - 85.96\%$ ) in the membrane and $75.57\% \pm 29.66$ (range $26.61\% - 99.77\%$ ) in the cytoplasm. In the membrane, the mean fluorescence intensity (MFI – absolute value) of NS4A was $30.23 \pm 9.03$ (range $19.11 - 43.71$ ). In the cytoplasm, the MFI was $63.32 \pm 32.41$ (range, 21.29-107.5). The expression index NS4A in the cytoplasm was 2.09. Confocal images show the expression of NS4A in yellow, CD61 in red, 7AAD in blue, and overlay markers in the sample. MK: megakaryocyte; MFI: mean fluorescence intensity.
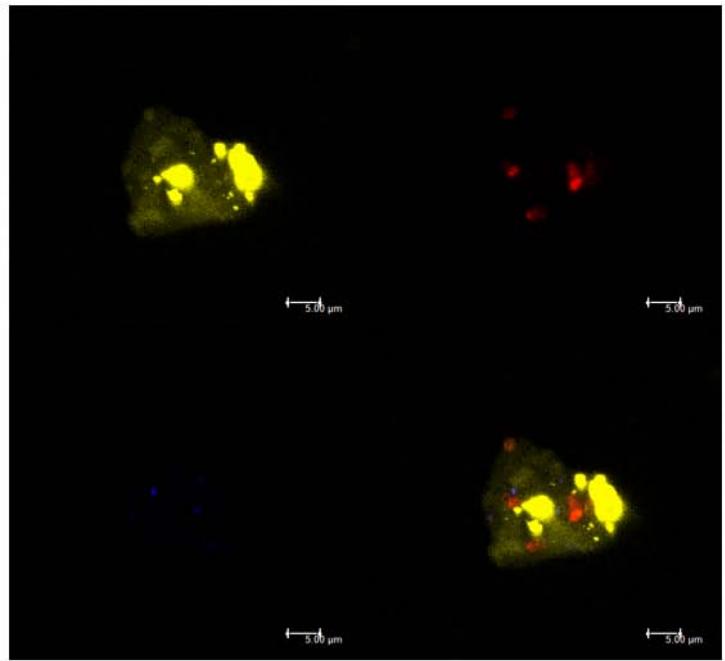
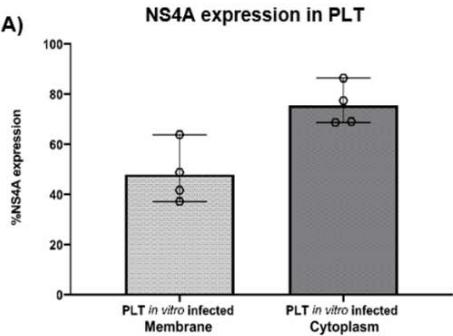
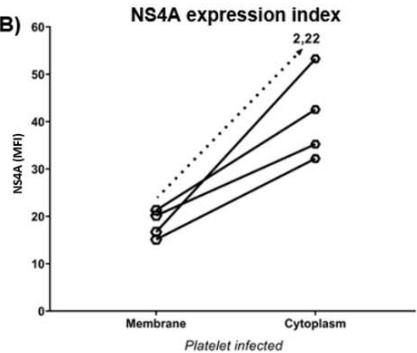
In vitro infected platelets expressed NS4A on both the membrane $(47.89\% \pm 11.66$ range $37.22\% - 63.85\%)$ and cytoplasm $(75.41\% \pm 8.36$ range $68.73\% - 86.43\%)$, similar to what was observed in megakaryocytes (Figure 2-A). The NS4A MFI was higher inside the cell $(40.82 \pm 9.37$ range 32.2-53.28) than in the membrane $(18.32\pm 2.89$ range 15.12-21.29) (Figure 2-B). The expression index NS4A in the cytoplasm was 2.22. Confocal images of in vitro infected platelets showed co-expression of NS4A and CD61 on the surface and in the cytoplasm of the cells (Figure 2C).
 A)
 B)
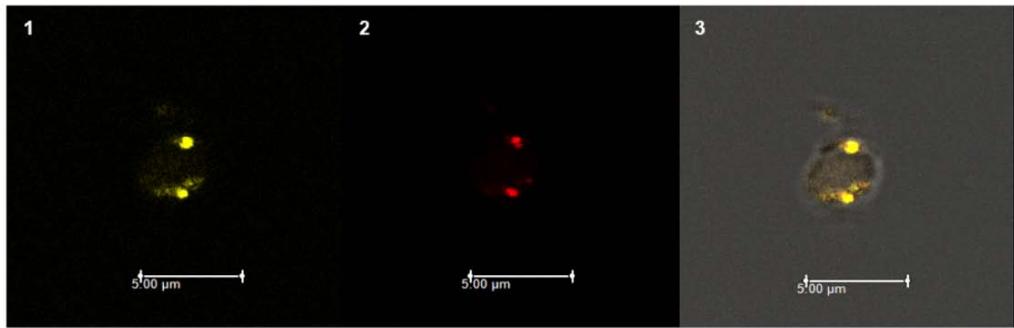 C) Figure 2: Platelets infected with HCV were evaluated in vitro using flow cytometry and confocal microscopy. The percentage expression of NS4A (A) was observed in the membrane $(47.89\% \pm 11.66$, range $37.22\% - 63.85\%)$ and in the cytoplasm $(75.41\% \pm 8.36$, range $68.73\% - 86.43\%)$. The median fluorescence intensity of NS4A (B) in the membrane $(18.32 \pm 2.89$, range 15.12-21.29) and in the cytoplasm $(40.82 \pm 9.37$, range 32.2-53.28) indicated an increase ratio of 2.22. The confocal images (C) obtained show the expression of NS4A in yellow (1), CD61 in red (2), and the overlay of both (3). PLT: platelet; MFI: mean fluorescence intensity.
To analyze viral expression in platelets in vivo, we evaluated samples from HCV-positive patients with detectable viral loads $(n = 2)$. Patient 1 had an HCV genotype 3 and a viral load of 17.051 IU/mL (4.23 log). Patient 2 had genotype 1B with a viral load of 3.243.950 UI/mL (6.51 log). According to the in vitro findings, platelets express higher viral concentrations in the cytoplasm compared to the cell surface. The results of the histological evaluation demonstrated the co-expression of CD61 and NS4A in liver tissue from HCV+ patients (Figure 3), reinforcing the idea that platelets can carry the virus and serve as extrahepatic reservoirs.
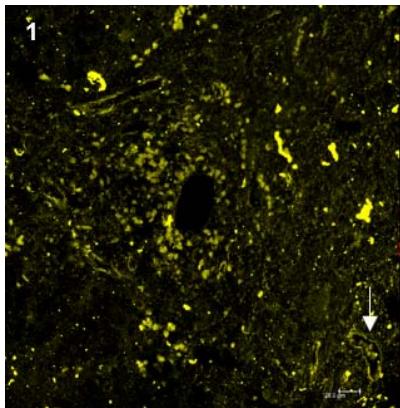
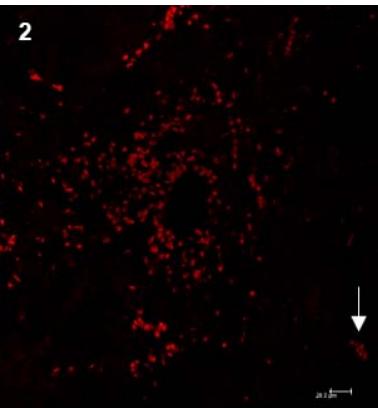
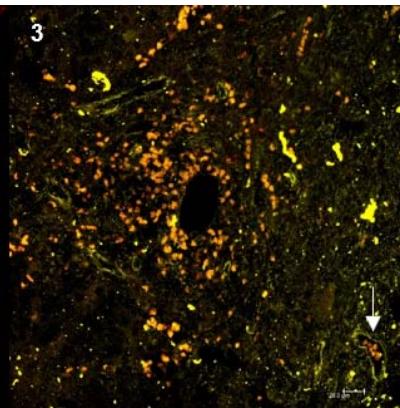 Figure 3: Viral expression in platelets in liver fragments from HCV patients: histological analysis using fluorescent markers. 1) Expression of NS4A on platelet surface; 2) CD61, platelet marker; 3) Overlay of NS4A and CD61. The arrows indicate HCV+ platelets in the blood vessels.
b) Megakaryocytes express claudin-1 and CD81, whereas platelets express only claudin-1.
The main entry receptors associated with HCV infection are claudin-1 and CD81 [32,33]. In this study, both HCV-infected and uninfected megakaryocytes showed expression of these receptors. Claudin-1 was expressed in $79.47\% \pm 10.42$ of HCV-infected megakaryocytes (with an MFI of $27.97 \pm 5.97$ ) and $69.75\% \pm 26.49$ of uninfected megakaryocytes (MFI $21.24 \pm 7.04$ ). CD81 was expressed in $61.10\% \pm 14.05$ of HCV-infected megakaryocytes (MFI $220.89 \pm 193.95$ ), and $67.56\% \pm 13.39$ of uninfected megakaryocytes (MFI $105.47 \pm 86.46$ ). It is important to emphasize that the in vitro infection process did not induce loss of expression of these receptors, as there was no significant difference between uninfected and infected megakaryocytes (Figure 4A, 4B, 4D). However, higher rates of CD81 (MFI) receptors in the presence of the virus suggest the involvement of immunomodulatory mechanisms.
Platelets expressed claudin-1 and were negative for CD81, confirming the expected phenotype. For in vitro infected platelets, the mean claudin-1 expression was $47.16\% \pm 10.36$ and MFI was $17.65 \pm 4.37$, whereas claudin-1 expression in uninfected platelets was $44.83\% \pm 8.26$ and MFI was $16.36 \pm 1.27$ (Figure 4C).
CD81, also known as TAPA-1, has four transmembrane domains that play a role in structural adhesion, activation, proliferation, and cell differentiation. According to microscopy images, the membranes of megakaryocytes expressed CD81, highlighting some cell regions with clusters forming structures known as tetraspanin-enriched microdomains. These microdomains are highly dynamic areas on the cell surface that facilitate interaction with other membrane components, crucial for the entry of HCV (Figure 4D-3) [33-34].
Unlike megakaryocytes, platelets expressed claudin-1 and are negative for CD81. For platelets infected in vitro with HCV, claudin-1 expression was $47.16\% \pm 10.36\%$ (range $37.08\% -57.89\%$ ) and MFI was $17.65 \pm 4.37$ (range 13.7-23.5). Uninfected platelets (exposed to HCV-negative plasma) showed a claudin-1 expression of $44.83\% \pm 8.26\%$ (range $33.07\% -52.42\%$ ) and an MFI of $16.36 \pm 1.27$ (range 14.86-17.94) (Figure 4C). There was no significant difference in claudin-1 expression between HCV-infected and uninfected platelets. Confocal analysis showed a homogeneous distribution on the platelet surface (data not shown).
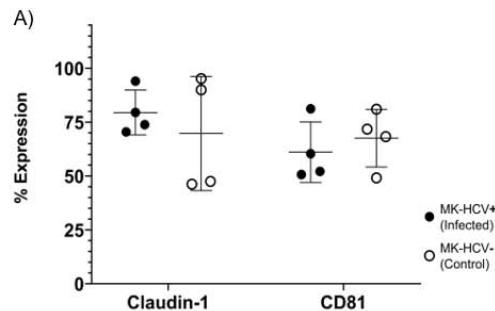
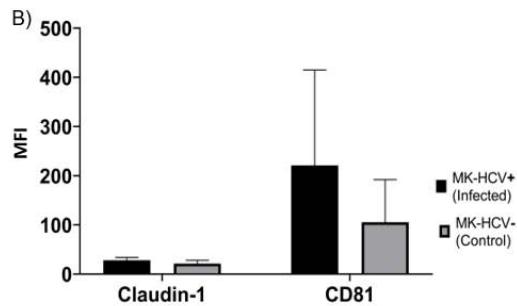
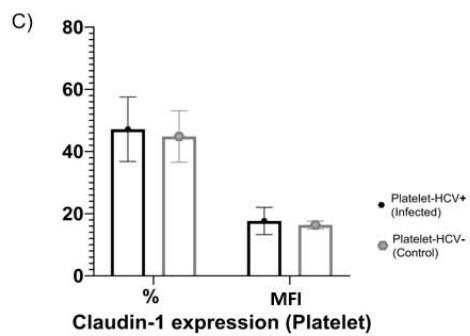
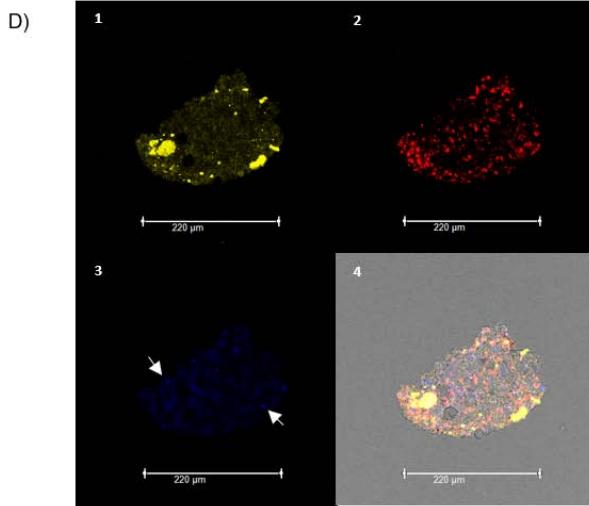 Figure 4: Expression of Claudin-1 and CD81 in megakaryocytes and platelets. (A) Percentage expression of claudin-1 (left) in infected megakaryocytes $79.47\% \pm 10.42\%$ (range $70.46\% - 94.07\%$ ) and uninfected megakaryocytes (control) $69.75\% \pm 26.49\%$ (range $46.26\% - 95.24\%$ ) and CD81 (right) in infected megakaryocytes $61.10\% \pm 14.05\%$
(range 52.15%-81.01%) and uninfected megakaryocytes (control) $67.56\% \pm 13.39\%$ (range 49.15%-81.01%); (B) MFI Claudin-1 expression in infected megakaryocytes $27.97 \pm 5.97$ (range 21.0-33.38), uninfected megakaryocytes $21.24 \pm 7.04$ (range 14.99-28.39) and MFI CD81 $220.89 \pm 193.95$ (range 41.42-403.15) and uninfected megakaryocytes (control) $105.47 \pm 86.46$ (range $36.85 \pm 231.86$ ) (C) Claudin-1 expression on platelets; (D) Confocal images of megakaryocytes: 1- Claudin-1 (yellow); 2- CD61 (red); 3- CD81 (blue) and tetraspanin microdomain (white arrows); 4- overlapping markers. MK: megakaryocyte; MFI: mean fluorescence intensity; HCV: hepatitis C virus.
## IV. DISCUSSION
HCV was detected in both the surface and the interior of megakaryocytes, with clear areas of higher virus concentration. These findings confirm that megakaryocytes are susceptible to HCV. They expressed high levels of CD81, the major viral entry receptor. Furthermore, molecular analyses have shown that megakaryocytes can support viral replication by detecting the complementary strand of viral RNA. This corroborates previous studies [2,22,35,36] and characterizes the virus-permissive cell function.
Our results regarding viral expression on the surface and cytoplasm of megakaryocytes strengthen our hypothesis that HCV can infect platelet precursors. This suggests that the virus may be transmitted during the differentiation of megakaryocytes into proplatelets, generating infected young platelets, such as swine flu [37]. Our results align with those of Li et al., who studied thrombocytopenia in patients with hepatitis C [2]. They demonstrated that megakaryocytes from a patient with megakaryoblastic leukemia in vitro infected with HCV were permissive to the virus [2]. Moreover, electron microscopy analyses revealed that the virus tends to accumulate around the Golgi complex and vesicles present in megakaryocytes [2]. Our observations using confocal microscopy, which identified areas of higher virus concentration in megakaryoblastic, corroborate these findings.
Several cell lineages, including hepatocytes and megakaryocytes, present CD81 receptors. These molecules are distinguished from other transmembrane proteins by the presence of conserved regions in their extracellular domains. These regions enable CD81 receptors to associate with other proteins such as integrins, signaling molecules, and homo- and heterodimer proteins to form tetraspanin-enriched microdomains [38,39,40]. These microdomains are coordinator molecules required for molecular trafficking, cell-cell fusion, motility, and signaling. Pathogens such as HCV and HIV use them as a gateway into the cell, facilitating viral entry [38-34].
The presence of HCV in the bone marrow correlates with the level of circulating viremia. Bone marrow changes such as hypo- and hypercellularity of erythroid, lymphoid, and myeloid lineages are attributed to factors such as viral load, viral subtype, immune status or immune complex deposition [19,22,36]. Abou El Azm et al. reported a decrease in the megakaryocyte population, the appearance of micromegakaryocytes, and abnormalities in the proliferation and differentiation of hematopoietic stem cells [41]. These observations suggest that HCV can interact directly with these cell populations and contribute to peripheral thrombocytopenia [22]. Similarly, El-Barbary et al. (2010) found a decrease in megakaryocytic colony-forming units and thrombocytopenia in HCV+ patients [42]. Since we have shown that megakaryocytes are permissive for HCV infection, we can infer that the bone marrow may be the extrahepatic site of viral replication.
HCV infection impairs the functions of hepatocytes, the major producers and secretors of thrombopoietin, a growth factor involved in platelet control and formation. Thrombopoietin reduction directly affects megakaryocyte ploidy and development, reducing platelet production. The presence of viral RNA in megakaryoblasts and megakaryocytes further promotes thrombocytopenia. This can occur through cell death or via reduction of the thrombopoietin receptor c-Mpl (myeloproliferative leukemia protein) [2,43,44]. The interaction between megakaryocytes and HCV also facilitates the imbalance in circulating thrombopoietin levels due to the lack of consumer cells for this factor. Consequently, serum thrombopoietin levels are significantly higher in patients with chronic hepatitis C [42].
The presence of HCV in platelets was confirmed here and in previous in vitro and in vivo studies [14,24]. Furthermore, when analyzing the viral RNA in plasma and platelets of HCV-positive patients over 144 hours, there was a decrease in plasma viral concentration, while RNA levels in platelets remained the same [23]. This suggests that HCV can persist in the body for long periods when associated with platelets, which can be considered a viral reservoir. Here, we showed for the first time that platelets express HCV both on the surface and inside the cell. Since the virus does not express CD81, the major receptor involved in HCV infection [32,33,34,38,45,46], the primary question is how the virus infects the platelets.
The mechanism of HCV entry or interaction in platelets is still unknown. Studies have found that cells expressing CD81 (t-Molt4 cells) and those lacking this marker (pro-monocytic lineage U937 and platelets) exhibit similar binding rates to HCV [24]. This indicates that HCV can interact with cells in the absence of the CD81 receptor. HCV may interact with platelets through adsorption or association with adhesion molecules such as anti-human platelet antigen (HPA)-1 integrins [14,47]. This interaction could occur through platelet antigens such as HPA-5b and HPA-1B, as their abundance is altered in platelets from hepatitis C patients [14,48]. Another interaction pathway may be through the glycoprotein VI receptor [37]. This receptor is a type 1 membrane glycoprotein belonging to the immunoglobulin (Ig) family with two Ig-C2 domains expressed on the membrane surface of platelets and megakaryocytes. IgG molecules associated with HCV could bind to the Ig-C2 domain, promoting not only cell-cell interaction but also viral dissemination and persistence [14,37,49].
Caludin-1, a junctional protein located on the apical and basolateral surfaces of hepatocytes, is an important receptor for HCV entry into these cells. Therefore, we investigated whether platelets and megakaryocytes express this protein. Claudin-1 functions either directly by binding to CD81 or through cell-to-cell infection. Due to its cellular location, claudin-1 may be able to interact with HCV [50,51]. Our data showed that megakaryocyte cells express claudin-1 and CD81, whereas platelets express only claudin-1. Cells expressing claudin-1 but lacking CD81 are susceptible to HCV [32]. Blocking the HCV-claudin-1 interaction with monoclonal antibodies prevented viral infection in vivo and in vitro, further implicating claudin-1 in HCV infection [33].
Our findings indicate that claudin-1 expression in megakaryocytes is not uniform but rather concentrated in specific areas of the cell. HCV enters hepatocytes through the association between CD81 and claudin-1. This complex facilitates the formation of tetraspanin-enriched microdomains, which are concentrated in certain cell regions. Therefore, we inferred that a similar process of HCV entry might occur in megakaryocytes.
HCV infects hepatocytes either through cell-free particle diffusion followed by engagement with specific cellular receptors or via cell-to-cell direct transmission mediated by mechanisms not well defined yet [52]. Cell-to-cell infection involves direct viral transfer to neighboring cells, allowing the virus to evade immune system cells and antibodies designed to fight it. This strategy may contribute to viral persistence [46,53-55].
CD81, claudin-1, occludin and low-density lipoprotein receptors are cellular molecules involved in cell-to-cell viral transmission. Since claudin-1was found to be expressed by platelets even in the absence of CD81, this pathway may be involved in platelet infection.[56]. Histological analysis of liver tissue from HCV patients revealed an accumulation of HCV-infected platelets $\left(\mathrm{CD61} + \mathrm{NS4A}+\right)$ around blood vessels. The capillaries or vessels in the hepatic endothelium play a critical role in the exchange of macromolecules, solutes, and fluids from the blood and are also the entry point for pathogens into the liver. We believe that platelets can transport the virus across the endothelium to liver tissue, which could favor the interaction of the virus with different receptors, including CD81, claudin-1, and occludin, allowing viral internalization in hepatocytes [57].
## V. CONCLUSION
This study demonstrated the presence of HCV on the platelet surface and, for the first time, identified viral expression in the cytoplasm of these cells. Possible mechanisms of interaction between HCV and platelets include adsorption due to platelet morphology/ phenotype or interaction mediated by receptors such as claudin-1, even in the absence of CD81. Additionally, we demonstrated HCV on both the surface and inside of megakaryocytes, suggesting that platelets derived from infected megakaryocytes may enter the circulation with the virus.
Our findings contribute to understanding the pathophysiology of hepatitis C, especially the interaction of the virus with different cell types. The presence of HCV in megakaryocytes and platelets, either through direct viral interaction that induces apoptosis or by altering the maturation of progenitor cells, may be related to thrombocytopenia, an extrahepatic manifestation often observed in patients with chronic hepatitis C.
Author Contributions: Conceptualization, P.E.A.M., G.F.S., M.I.M.C.P., M.A.G., M.A.C.D., R.M.T.G.; methodology, C.M.W.; A.M.M.B., M.A.C.D., S.F.C., N.A.S.T.; writing—original draft preparation, M.A.G., C.M.W., A.M.M.B., G.T.V., G.F.S. Review and editing, C.M.W, N.A.S.T., S.F.C., M.A.G., R.M.T.G.; G.T.V. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement: This study was approved by Research Ethics Committee of Botucatu Medical School, UNESP (protocol 1.354.285).
Informed Consent Statement: Informed consent was obtained from all subjects involved in the study.
Data Availability Statement: All data related to this article are available within the manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
Abbreviation list:
CD81 - Cluster of differentiation 81
- CPDA - Cit Phosph Dextrose Adenine
- EDTA - Ethylenediaminetetraacetic acid
- HCV - Hepatitis C virus
- Ig - Immunoglobulin
- MFI - Mean fluorescence intensity
- RPMI - Roswell Park Memorial Institute Medium
Generating HTML Viewer...
References
56 Cites in Article
Julie Sheldon,Nathan Beach,Elena Moreno,Isabel Gallego,David Piñeiro,Encarnación Martínez-Salas,Josep Gregori,Josep Quer,Juan Esteban,Charles Rice,Esteban Domingo,Celia Perales (2014). Increased Replicative Fitness Can Lead to Decreased Drug Sensitivity of Hepatitis C Virus.
X Li,L Jeffers,C Garon,E Fischer,J Scheffel,B Moore,K Reddy,M Demedina,E Schiff (1999). Persistence of hepatitis C virus in a human megakaryoblastic leukaemia cell line.
W Organization,Hepatitis,Virus (2023). Hepatitis C virus (HCV) fact sheet.
Nicola Fletcher,Garrick Wilson,Jacinta Murray,Ke Hu,Andrew Lewis,Gary Reynolds,Zania Stamataki,Luke Meredith,Ian Rowe,Guangxiang Luo,Miguel Lopez–ramirez,Thomas Baumert,Babette Weksler,Pierre–olivier Couraud,Kwang Kim,Ignacio Romero,Catherine Jopling,Susan Morgello,Peter Balfe,Jane Mckeating (2012). Hepatitis C Virus Infects the Endothelial Cells of the Blood-Brain Barrier.
Giovanna Russelli,Paola Pizzillo,Gioacchin Iannolo,Floriana Barbera,Fabio Tuzzolino,Rosa Liotta,Mario Traina,Giovanni Vizzini,Bruno Gridelli,Ester Badami,Pier Conaldi (2017). HCV replication in gastrointestinal mucosa: Potential extra-hepatic viral reservoir and possible role in HCV infection recurrence after liver transplantation.
M Granato,C Zompetta,E Vescarelli,C Rizzello,A Cardi,S Valia,G Antonelli,C Marchese,M Torrisi,A Faggioni,M Cirone (2016). HCV derived from sera of HCV-infected patients induces pro-fibrotic effects in human primary fibroblasts by activating GLI2.
H Muller,E Pfaff,T Goeser,B Kallinowski,C Solbach,L Theilmann (1993). Peripheral blood leukocytes serve as a possible extrahepatic site for hepatitis C virus replication.
Maria‐cristina Navas,Anne Fuchs,Evelyne Schvoerer,Alain Bohbot,Anne‐marie Aubertin,Françoise Stoll‐keller (2002). Dendritic cell susceptibility to hepatitis C virus genotype 1 infection.
R Wang,P Bare,V De Giorgi,K Matsuura,K Salam,T Grandinetti,C Schechterly,H Alter,I Castillo,E Rodríguez-Iñigo,J Bartolomé,S De Lucas,N Ortíz-Movilla,J López-Alcorocho,M Pardo,V Carreño (2005). Hepatitis C virus replicates in peripheral blood mononuclear cells of patients with occult hepatitis C virus infection.
T Chang,K Young,Y Yang,H Lei,H Wu (1996). Hepatitis C virus RNA in peripheral blood mononuclear cells: Comparing acute and chronic hepatitis C virus infection.
K Salam,R Wang,T Grandinetti,V De Giorgi,H Alter,R Allison (2018). Binding of Free and Immune Complex-Associated Hepatitis C Virus to Erythrocytes Is Mediated by the Complement System.
Yasuteru Kondo,T Shimosegawa (2013). Direct effects of hepatitis C virus on the lymphoid cells.
Juliana Padovani,Silvia Corvino,Jan Drexler,Giovanni Silva,Maria Pardini,Rejane Grotto (2013). In vitro detection of hepatitis C virus in platelets from uninfected individuals exposed to the virus.
H Lerat,M ; Trabaud,O Vidalin,M Major,C; Trépo,G Inchauspé (1996). Specificdetectionofhepatitis C virusminusstrand RNA in hematopoieticcells.
Adilson De Almeida,Marilza Campos-De-Magalhães,Olívia De Melo Marçal,Carlos Brandão-Mello,Margareti Okawa,Rosane De Oliveira,Márcia Do Espírito-Santo,Clara Yoshida,Elisabeth Lampe (2004). Hepatitis C virus-associated thrombocytopenia: a controlled prospective, virological study.
Sumit Dahal,Smrity Upadhyay,Rashmi Banjade,Prajwal Dhakal,Navin Khanal,Vijaya Bhatt (2017). THROMBOCYTOPENIA IN PATIENTS WITH CHRONIC HEPATITIS C VIRUS INFECTION.
S Kedia,V Bhatt,S Rajan,P Tandra,R El Behery,M Akhtari (2014). Hematological and Musculoskeletal Manifestations of Systemic Lupus Erythematosus in A Patient with Chronic Hepatitis C Virus Infection.
Howard Liebman (2008). Viral-Associated Immune Thrombocytopenic Purpura.
Massimo Franchini,Dino Veneri,Giuseppe Lippi (2017). Thrombocytopenia and infections.
Shojiro Ichimata,Mikiko Kobayashi,Kohei Honda,Soichiro Shibata,Akihiro Matsumoto,Hiroyuki Kanno (2017). Acquired amegakaryocytic thrombocytopenia previously diagnosed as idiopathic thrombocytopenic purpura in a patient with hepatitis C virus infection.
J Ariede,M Pardini,G Silva,R Grotto (2015). Platelets can be a biological compartment for the Hepatitis C Virus.
Samir Hamaia,Chengyao Li,Jean-Pierre Allain (2001). The dynamics of hepatitis C virus binding to platelets and 2 mononuclear cell lines.
G Bordin,M Ballaré,P Zigrossi,M Bertoncelli,L Paccagnino,A Baroli,M Brambilla,A Monteverde,E Inglese (1995). A laboratory and thrombokinetic study of HCV-associated thrombocytopenia: a direct role of HCV in bone marrow exhaustion?.
A Tomer,P Friese,R Conklin,W Bales,L Archer,L Harker,S Burstein (1989). Flow cytometric analysis of megakaryocytes from patients with abnormal platelet counts.
T Ishibashi,Z Ruggeri,L Harker,S Burstein (1986). Separation of human megakaryocytes by state of differentiation on continuous gradients of Percoll: size and ploidy analysis of cells identified by monoclonal antibody to glycoprotein IIb/IIIa.
Robert Liwski,Anna Greenshields,David Conrad,Cathi Murphey,Robert Bray,Jorge Neumann,Howard Gebel (2009). Rapid optimized flow cytometric crossmatch (FCXM) assays: The Halifax and Halifaster protocols.
A Roder,C Vazquez,S Horner (2019). The acidic domain of the hepatitis C virus NS4A protein is required for viral assembly and envelopment through interactions with the viral E1 glycoprotein.
Fabrice Cognasse,Hind Hamzeh,Patricia Chavarin,Sophie Acquart,Christian Genin,Olivier Garraud (2005). Evidence of Toll‐like receptor molecules on human platelets.
Sergey Shiryaev,Elliot Thomsen,Piotr Cieplak,Eugene Chudin,Anton Cheltsov,Mark Chee,Igor Kozlov,Alex Strongin (2012). New Details of HCV NS3/4A Proteinase Functionality Revealed by a High-Throughput Cleavage Assay.
Yong-Zhe Zhu,X Qian,P Zhao,Z Qi (2014). How hepatitis C virus invades hepatocytes: The mystery of viral entry.
L Fénéant,S Levy,L Cocquerel (2014). CD81 and hepatitis C virus (HCV) infection.
Selma Dahmane,Eric Rubinstein,Pierre-Emmanuel Milhiet (2014). Viruses and Tetraspanins: Lessons from Single Molecule Approaches.
A Manzin,M Candela,S Paolucci,M Caniglia,A Gabrielli,M Clementi (1994). Presence of hepatitis C virus (HCV) genomic RNA and viral replicative intermediates in bone marrow and peripheral blood mononuclear cells from HCV-infected patients.
Marek Radkowski,Joanna Kubicka,Elzbieta Kisiel,Janusz Cianciara,Marek Nowicki,Jorge Rakela,Tomasz Laskus (2000). Detection of active hepatitis C virus and hepatitis G virus/GB virus C replication in bone marrow in human subjects.
Astrid Zahn,Nicola Jennings,Willem Ouwehand,Jean-Pierre Allain (2006). Hepatitis C virus interacts with human platelet glycoprotein VI.
Noha Hassuna,Peter Monk,Gregory Moseley,Lynda Partridge (2009). Strategies for Targeting Tetraspanin Proteins.
Stephen Fitter,Paul Sincock,Corina Jolliffe,Leonie Ashman (1999). Transmembrane 4 superfamily protein CD151 (PETA-3) associates with β1 and αIIbβ3 integrins in haemopoietic cell lines and modulates cell–cell adhesion.
C Boucheix,E Rubinstein (2001). Tetraspanins.
A Abou El Azm,H El-Bate,L Abo-Ali,N Mansour,H Ghoraba,M Salem (2012). Correlation of viral load with bone marrow and hematological changes in pale patients with chronic hepatitis C virus.
Magdy El Barbary,Alaa Saad,Fadia Attia,Magda Mandour,Mohamed Haidara,Mohammad Dallak,Esma Isenovic (2010). Thrombocytopenia in Patients With Chronic Hepatitis C: A Possible Role of HCV on Platelet Progenitor Cell Maturation.
N Afdhal,J Mchutchison (2007). Review article: pharmacological approaches for the treatment of thrombocytopenia in patients with chronic liver disease and hepatitis C infection.
Alice Assinger (2014). Platelets and Infection – An Emerging Role of Platelets in Viral Infection.
Yong-Zhe Zhu,Yuan Luo,Ming-Mei Cao,Yuan Liu,Xiao-Qing Liu,Wen Wang,Da-Ge Wu,Mo Guan,Qing-Qiang Xu,Hao Ren,Ping Zhao,Zhong-Tian Qi (2012). Significance of palmitoylation of CD81 on its association with tetraspanin-enriched microdomains and mediating hepatitis C virus cell entry.
R Tawar,C Colpitts,J Lupberger,H El-Saghire,M Zeisel,T Baumert (2015). Claudins and pathogenesis of viral infection.
Quentin Sattentau (2008). Avoiding the void: cell-to-cell spread of human viruses.
Camila Verdichio‐moraes,Cecília Toralles‐pereira,Rejane Grotto,Giovanni Silva,Maria Pardini (2009). Allelic frequencies of HPA‐1 to 5 human platelet antigens in patients infected with hepatitis C virus.
Galit Alter,Jessica Malenfant,Marcus Altfeld (2004). CD107a as a functional marker for the identification of natural killer cell activity.
Ágnes Holczbauer,Benedek Gyöngyösi,Gábor Lotz,Péter Törzsök,Pál Kaposi-Novák,Attila Szijártó,Péter Tátrai,Péter Kupcsulik,Zsuzsa Schaff,András Kiss (2014). Increased Expression of Claudin-1 and Claudin-7 in Liver Cirrhosis and Hepatocellular Carcinoma.
Jesús Torres-Flores,Carlos Arias (2015). Tight Junctions Go Viral!.
Virgínia Gondar,Francisca Molina-Jiménez,Takayuki Hishiki,Luisa García-Buey,George Koutsoudakis,Kunitada Shimotohno,Ignacio Benedicto,Pedro Majano (2015). Apolipoprotein E, but Not Apolipoprotein B, Is Essential for Efficient Cell-to-Cell Transmission of Hepatitis C Virus.
J Witteveldt,M Evans,J Bitzegeio,G Koutsoudakis,A Owsianka,A Angus,Z-Y Keck,S Foung,T Pietschmann,C Rice,A Patel (2009). CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells.
Isabel Fofana,Samira Fafi–kremer,Patric Carolla,Catherine Fauvelle,Muhammad Zahid,Marine Turek,Laura Heydmann,Karine Cury,Juliette Hayer,Christophe Combet,François–loïc Cosset,Thomas Pietschmann,Marie–sophie Hiet,Ralf Bartenschlager,François Habersetzer,Michel Doffoël,Zhen–yong Keck,Steven Foung,Mirjam Zeisel,Françoise Stoll–keller,Thomas Baumert (2012). Mutations That Alter Use of Hepatitis C Virus Cell Entry Factors Mediate Escape From Neutralizing Antibodies.
Frederik Graw,Danyelle Martin,Alan Perelson,Susan Uprichard,Harel Dahari (2015). Quantification of Hepatitis C Virus Cell-to-Cell Spread Using a Stochastic Modeling Approach.
Huahao Fan,Luhua Qiao,Kyung-Don Kang,Junfen Fan,Wensheng Wei,Guangxiang Luo (2017). Attachment and Postattachment Receptors Important for Hepatitis C Virus Infection and Cell-to-Cell Transmission.
Birke Bartosch,Jean Dubuisson (2010). Recent Advances in Hepatitis C Virus Cell Entry.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Rejane Maria Tommasini Grotto. 2026. \u201cHepatitis C Virus Infected Human Megakaryocytes and Platelets: Intra- and Extracellular Evaluation\u201d. Global Journal of Medical Research - C: Microbiology & Pathology GJMR-C Volume 24 (GJMR Volume 24 Issue C1): .
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
This study aimed to determine whether the hepatitis C virus (HCV) infects human megakaryocytes and platelets and to measure the expression of receptors involved in virus-cell interaction. Platelets from healthy donors were infected with HCV in vitro and analyzed for viral expression and the receptors claudin-1 and cluster of differentiation 81 (CD81). HCV was detected on the surface and cytoplasm of both cells; cytoplasmic expression was higher compared to the surface. Platelets presented a claudin-1+/CD81-phenotype, and megakaryocytes showed aclaudin-1+/CD81+ phenotype. We conclude that megakaryocytes and platelets are susceptible to HCV infection, regardless of CD81 expression, and megakaryocytes may serve as possible sites of viral replication.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.