Breast cancer remains the most deadly cancer in women worldwide. It is a highly heterogeneous disease group, both biologically and molecularly. Mammary carcinogenesis is a multi-stage, complex and progressive process, involving the accumulation of several genetic and epigenetic abnormalities in oncogenes and suppressor genes. These abnormalities lead to activation or inhibition of various molecules involved in cellular and molecular signaling pathways, thus altering stem cell proliferation, differentiation and cell death. Patients with certain constitutional genetic abnormalities are a sub-population at high risk of accumulating several molecular abnormalities at an early stage, and of developing more invasive breast cancers. Understanding the molecular pathogenesis of breast cancer is an essential step towards distinguishing molecular subtypes with different prognostic and therapeutic implications. This review provides a synthesis of the major molecular abnormalities found in breast cancer, focusing on molecules that are considered in the literature as prognostic or theranostic markers.
## I. INTRODUCTION
Breast cancer is the leading cause of cancer-related death in women. Over $80\%$ of cases occur sporadically, underlining the importance of somatic abnormalities, which involve several risk factors related to the patient's lifestyle. Only $15 - 20\%$ of breast cancers occur in a context suggestive of hereditary transmission of a mutation in a gene predisposing to the development of cancer. Mutation of the BRCA1 or BRCA2 genes is found in only $25\%$ of cases. Thanks to extraordinary advances in molecular biology over the last few decades, our understanding of the cellular and molecular basis of cancer has broadened considerably. In this summary, the authors present a review of the signaling pathways most frequently involved in the carcinogenesis of sporadic forms of breast cancer, as well as the main predisposition genes found in hereditary forms.
## II. CELLS OF ORIGIN OF BREAST CANCER
The determination of the origin of breast cancer cells has been elucidated in the light of the understanding of the normal cell hierarchy. Mammary stem cells (MSCs), constituting a very small proportion of mammary gland cells, are undifferentiated and can produce new MSCs by self-renewal and give rise to a variety of differentiated cells by symmetrical and asymmetrical divisions. Asymmetric divisions give rise to ductular, alveolar and myoepithelial progenitor cells. Several processes of division with differentiation are initiated to give rise to mature ductular, alveolar and myoepithelial cells. In the healthy body, MSCs are involved in responding to cellular needs during reproductive life. This is thanks to a close interaction with their specific cellular microenvironment, known as the mammary stem cell niche [1].
Comparison of the established molecular characteristics of normal breast epithelial subpopulations with those of different breast cancer subtypes (luminal A, luminal B, HER2-positive, claudin-low, and basal-like), has provided an important framework for understanding the cellular origins of this cancer, both sporadic and hereditary. Cancer subtypes appear to aggregate along the hierarchy of normal cellular differentiation, starting with claudin-low undifferentiated tumors, followed by basal-like tumors, HER2 tumors and finally luminal A and B tumor subtypes [2]. On a molecular level, MSCs are similar to the low-claudin cancer subtype. The luminal progenitor subset has a molecular profile very similar to that of the tumor cells found in the basal-like subtype. The HER2, luminal A and luminal B subtypes reflect different cell types within the luminal lineage. The molecular profile of luminal A tumors is closest to that of mature luminal cells [2].
With regard to familial breast cancer, it was initially proposed that the mammary stem cell resident in the basal layer of the mammary epithelium was the "cell of origin" of BRCA1-mutated basal-like tumors. This proposal was mainly based on histological studies that noted similarities between basal-like tumors and basal epithelial cells, i.e. the expression of basal cytokeratins and the absence of hormone receptor expression. Early work suggested that cells carrying the BRCA1 gene mutation show disturbed differentiation. Currently, analysis of the pre-neoplastic tissue of BRCA1 mutation carriers has revealed a target population predisposed to neoplastic transformation: an enlarged population of aberrant luminal progenitor cells. These cells show the greatest molecular similarity to normal luminal progenitor cells [2]. An experiment was carried out in 2011 involving transduction of human mammary epithelial cells with a cocktail of potent oncogenic lentiviruses, followed by implantation in humanized mammary adipose tissue. Results showed that BRCA1-
Mutated luminal cells were more susceptible to malignant transformation than basal cells, and produced predominantly basal-like tumors[3]. Luminal progenitor cells are reprogrammed to acquire basal-like characteristics. This is mediated in part by the BRCA1-regulated SLUG transcription factor. This epithelial-mesenchymal transition factor is overexpressed in tissues with a BRCA1 mutation and thus blocks luminal cell differentiation, directing the cells towards a basal fate[3].
Cell fate decisions along the mammary epithelial hierarchy may not be strictly unidirectional. Increasing evidence suggests that the dedifferentiation process can occur under non-physiological conditions. Luminal epithelial cells can convert to basal-like cells upon oncogenic stress in vivo, and induction of a P53 protein mutation in luminal cells produces tumors with basal-like features (Figure1). These data reflect the inherent plasticity of the mammary luminal compartment during carcinogenesis [4].
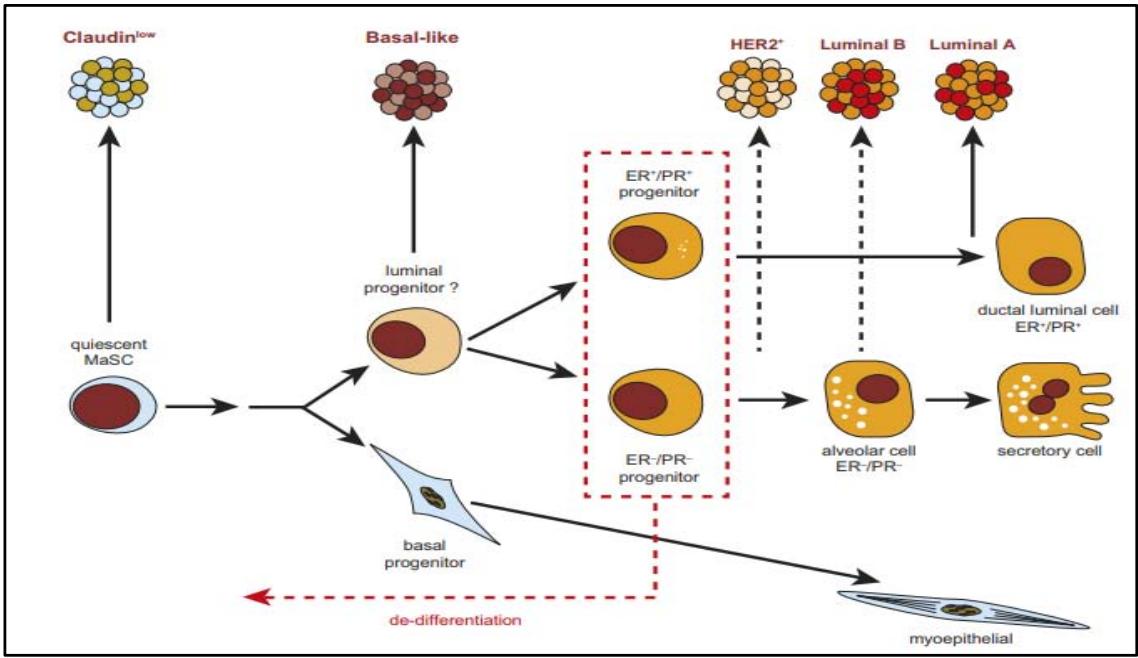 Figure 1: Schematic model of the relationships between cells of the breast epithelial hierarchy and breast tumour subtypes. Luminal progenitors also show marked de differentiation during oncogenesis. [2].
## III. SIGNALING PATHWAYS INVOLVED IN BREAST CANCER DEVELOPMENT AND PROGRESSION
Cancer is caused by genetic and epigenetic alterations that disrupt cell signaling pathways. In this way, the tumor cell manages to escape the control mechanisms of proliferation, survival and migration. The main alterations in signaling pathways found in breast cancer stem cells are as follows:
### A/Signaling pathways involving estrogen receptors
The estrogen receptor signalling pathway is the most common pathway in breast cancer. It involves estrogen ligands, which are transcription factors that activate or repress the expression of target genes upon receptor binding. There are two types of estrogen receptor: G protein-coupled membrane receptors and nuclear ERα, ERβ receptors. Normal breast tissue frequently expresses ERβ-type nuclear receptors. They are most often active as dimers. Although located on two different loci, ERα encoded by ESR1 located on the long arm of chromosome 6 and ERβ encoded by ESR2 located on the long arm of chromosome 14, these two receptors share common structural features.
In fact, ER $\alpha$ and ER $\beta$ are organized into 5 functional domains, designated A to F from the N-terminus to the C-terminus (Figure 2):
- The C domain includes the DNA Binding Domain (DBD), which binds to chromatin response elements known as EREs (Estrogen Responsive Elements). EREs are DNA sequences located at the promoter of target genes.
- The D domain, also known as the Hinge domain, ensures the receptor's flexibility at DNA level. It also contains a nuclear localization signal.
- The larger E domain contains the ligand-binding domain (LBD). It also ensures ligand-dependent
transactivation of transcription (Activation Function 2 AF-2).
- The F domain, located at the C-terminus of the receptor, is still largely unknown. However, it could modulate ER $\alpha$ transcriptional activity and protein-protein interactions, notably with SRC-1 (Steroid Receptor Coactivator-1).
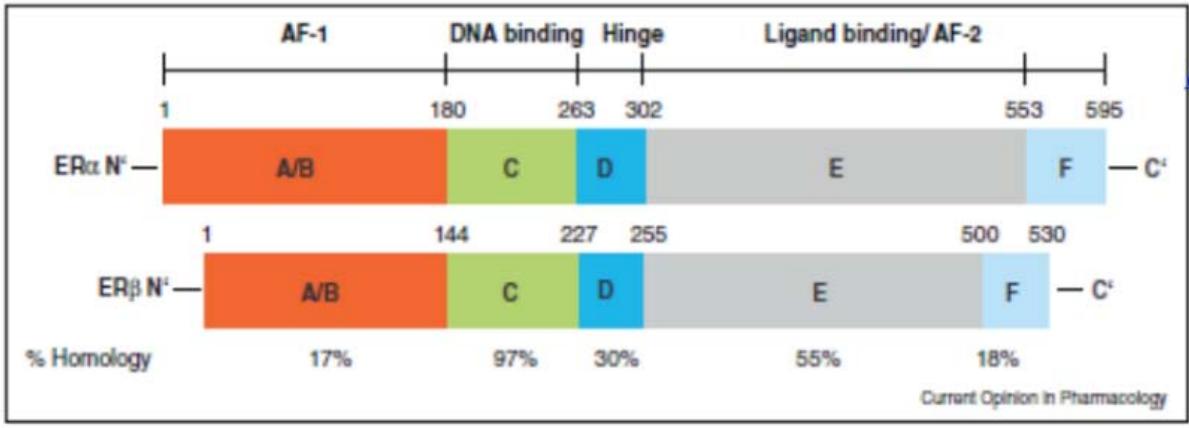 Figure 2: The functional domains of oestrogen receptors, numbered A to F (Pr Jacqueline LEHMANN CHE). Panel label: ER.
$\alpha$ and ER $\beta$ share $98\%$ homology in their DNA-binding domains (DBD) (Pace et al., 1997), while they differ widely in their transcription activation domains (less than $15\%$ homology in their N-terminal domains). Indeed, the N-terminal part of ER $\beta$ is about 40 amino acids shorter than that of ER $\alpha$. Its AF-1 transcriptional activity is thus considerably reduced. As a result, ER $\alpha$ and ER $\beta$ recruit co-activator proteins differently, modifying their specific transcriptional effects.
ER $\alpha$ and ER $\beta$ share a $55 - 59\%$ homology in their LBD (ligand-binding domain), which influences their affinity for their ligands. Moreover, ER $\alpha$ is associated with cell proliferation, whereas ER $\beta$ is thought to play an antiproliferative role [5].
#### 1/ The genomic pathway
#### - Direct genomic pathway
The direct genomic pathway is activated when estrogen (E2) binds to its receptor, resulting in a conformational change. The conformational change of estrogen receptors facilitates the association and dissociation of enzymatic co-regulators. These proteins are either histone acetyltransferases or histone methyltransferases, or ATPase complexes such as SWI/SNF, which participate in chromatin remodeling. The receptor is then translocated into the nucleus and binds to specific DNA sequences, the EREs (Estrogen Responsive Elements), and activates the transcription of target genes via its AF-1 and AF-2 domains. A transcriptional machinery containing RNA polymerase II and TBP (TATA binding protein) is recruited to initiate transcription.
This stage requires the intervention of pioneering factors known as co-activators, including the main ones SRC, GATA3 and the FOXA1 protein. These proteins, especially FOXA1, play an important role in ER $\alpha$ binding to chromatin and activate transcription of genes involved in cell cycle progression, notably CCND1, which codes for cyclin D1 [6]. The latter is an important activator of cyclin-dependent kinases (CDK) 4 and 6, which coordinate cell cycle transition from G1 to S phase in many cancer cells (Figure 3). In breast tumor lines, it has been shown that ER $\alpha$ binds to chromatin even in the absence of estrogen, but in a FOXA1-dependent manner [6] The feedback loop between ER $\alpha$ and cyclin D1 may explain the mechanism of resistance to antiestrogen therapy, and justifies the use of kinase 4 and 6 inhibitors in combination with hormone therapy. [1].
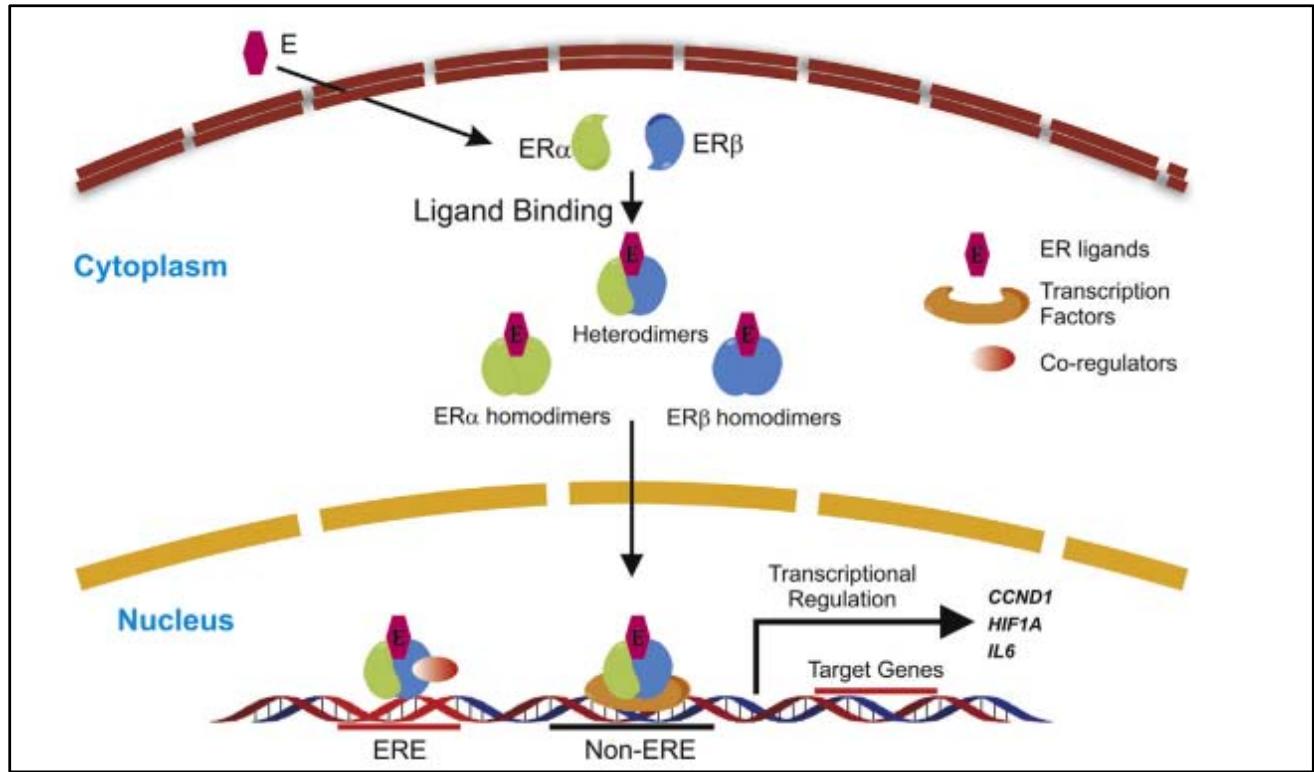 Figure 3: Direct genomic signalling pathway of oestrogen receptors ER: Nuclear hormone receptors form homo- or heterodimers on ligand binding and move into the nucleus for transcriptional regulation. ER dimersbind to the ERE region of target genes and recruitco-regulators such as FOXA 1 [1].
There are several isoforms with paradoxical effects in the regulation of ER $\alpha$ signaling. ERa36, the isoform most frequently found in metastatic breast cancer, is not blocked by tamoxifen. On the contrary, once bound to ER $\alpha$ 36, it promotes disease progression. The ER $\alpha$ 36 isoform accounts for $70\%$ of resistance to hormone therapy [7].
#### The indirect genomic pathway
A proportion of estrogen receptors activate the transcription of target genes by binding to other transcription factors already present on chromatin, without binding to ERE (Estrogen Responsive Elements) sites. This pathway is also known as the ERE-independent genomic pathway. In this case, the estrogen receptor acts as a transcriptional cofactor. The indirect genomic pathway is reserved for Erα, ERb have no coactivating activity. Several genes are activated by estrogen (E2) via the interaction of the estrogen nuclear receptor with transcription factors such as SP-1 (Specificity Protein 1), NF-κB (Nuclear Factor-κB), GATA1, STAT5 (Signal Transducer and Activator of Transcription 5) and AP-1 (Activating Protein 1). Genes activated in this way include those encoding IGF-1 (Insulin Growth Factor 1), cyclin D1, C-Myc protein, Bcl2 [8].
The activity of nuclear estrogen receptors can be modulated by signals other than estrogen. This is made possible by the ligand-independent AF-1 transactivation domain. Phosphorylation of estrogen receptors on serine 118 residues enables ER $\alpha$ to be localized at several target gene promoters to activate their transcription. This phosphorylation is induced either by type A and C protein kinases (PKA, PKC), cell cycle regulators, neurotransmitters, or growth factors such as EGF (Epidermal Growth Factor), IGF-1 (Insulin-like Growth Factor), TGF $\beta$ (Transforming Growth Factor) [9]. 2/ the non-genomic route
The idea of the existence of a non-genomic estrogen-mediated pathway has been suggested since 1977, given that some estrogen-induced changes are too rapid to be mediated by the genomic pathway. A subset of ERα, localized to the plasma membrane, is involved in extranuclear signaling cascades. One of the most well-documented interactions is the ERα/Src interaction, which occurs rapidly after estrogen stimulation, leading to Src activation. Src is a tyrosine kinase which in turn phosphorylates RAS. This activation cascade induces the activation of MEK, which directly phosphorylates ERK1/2. Phosphorylated ERK1/2 migrates to the nucleus to activate genes such as CCND1 [8].
In addition to all the above-mentioned pathways, it has been shown that in the presence of estrogen, ERα rapidly interacts with the regulatory subunit of PI3 kinase (PI3K), enabling cells to enter the S phase of the cell cycle, and activate Cyclin D1. In 2012 a team studied the formation of the ERα/PI3K/Src complex in a cohort of 175 breast tumors. It showed that activation of this complex was correlated with a poor prognosis and a low relapse-free survival rate [10].
#### B/ HER2 signaling pathways
HER2 is a proto-oncogene corresponding to a transmembrane protein encoded by the ERBB2 gene, located on the long arm of chromosome 17. HER2 belongs to the epidermal growth factor receptor tyrosine kinase family, which comprises 4 subtypes (EGFR)/HER1, HER2, HER3 and HER4. It controls cell growth, survival, differentiation and migration [11].
The molecular structure of the EGFR family consists of a large extracellular region, a single-span transmembrane (TM) domain, an intracellular juxtamembrane (JM) region, a tyrosine kinase domain and a C-terminal regulatory region. HER3 is the only tyrosine kinase-deficient receptor, which is why it assumes no signal transduction. The ligand for HER2 has not yet been identified. HER2 undergoes ligand-independent heterodimerization with the other 3 members of the EGFR family. At high HER2 concentrations, HER2 may undergo homodimerization due to its constitutively active conformation [12].
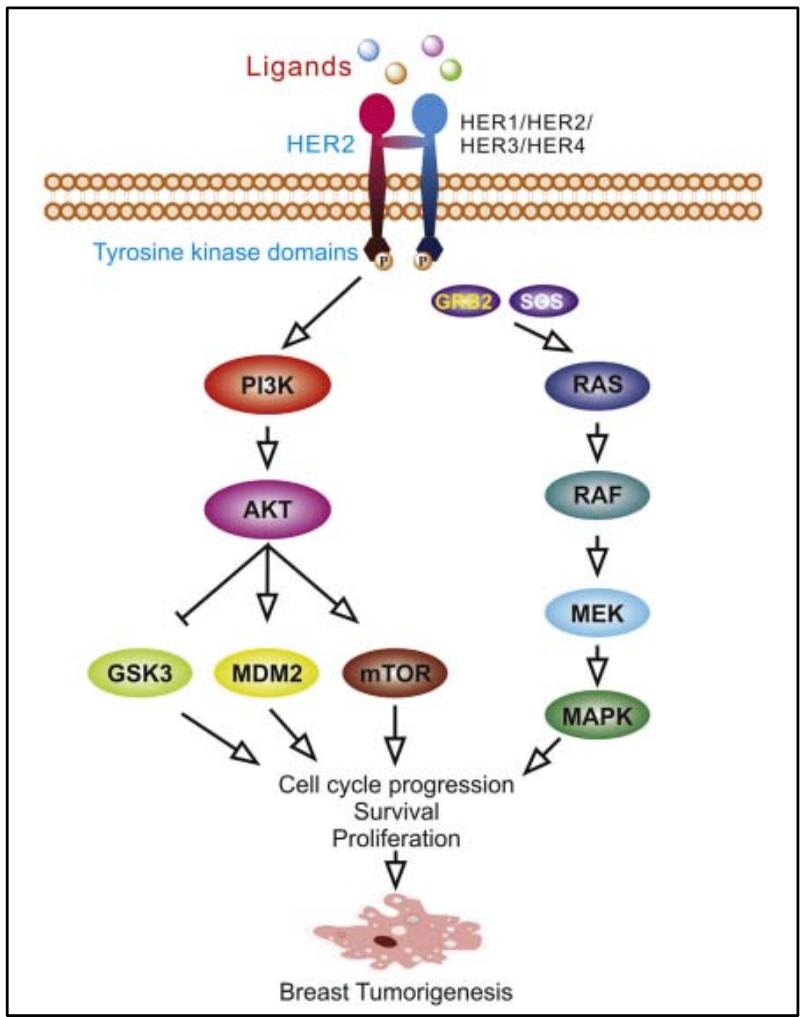
The formation of homodimers and heterodimers brings the intracellular domains closer together, resulting in asymmetric interaction of the intracellular kinase domain between the amino-terminal lobe of one tyrosine kinase and the carboxy-terminal lobe of the other, and promoting autophosphorylation of the tyrosine kinase domains. Several signalling pathways are then activated, including PI3K/Akt, MAPK, PLC $\gamma$, ERK1/2, JAK/STAT. MAPK and PI3K/Akt are the two main pathways activated by the EGFR family, in particular the HER2 heterodimer (Figure 4). The activated MAPK pathway promotes transcription of related genes, subsequently enhancing cancer cell proliferation, migration, differentiation, angiogenesis and drug resistance. In the PI3K/Akt pathway, phosphorylated Akt acts on a range of transcription factors including MDM2, mTOR, p27, GSK3β, BAD, NF-κB, FKHR, enhancing proliferation, survival and suppressing apoptosis [12].
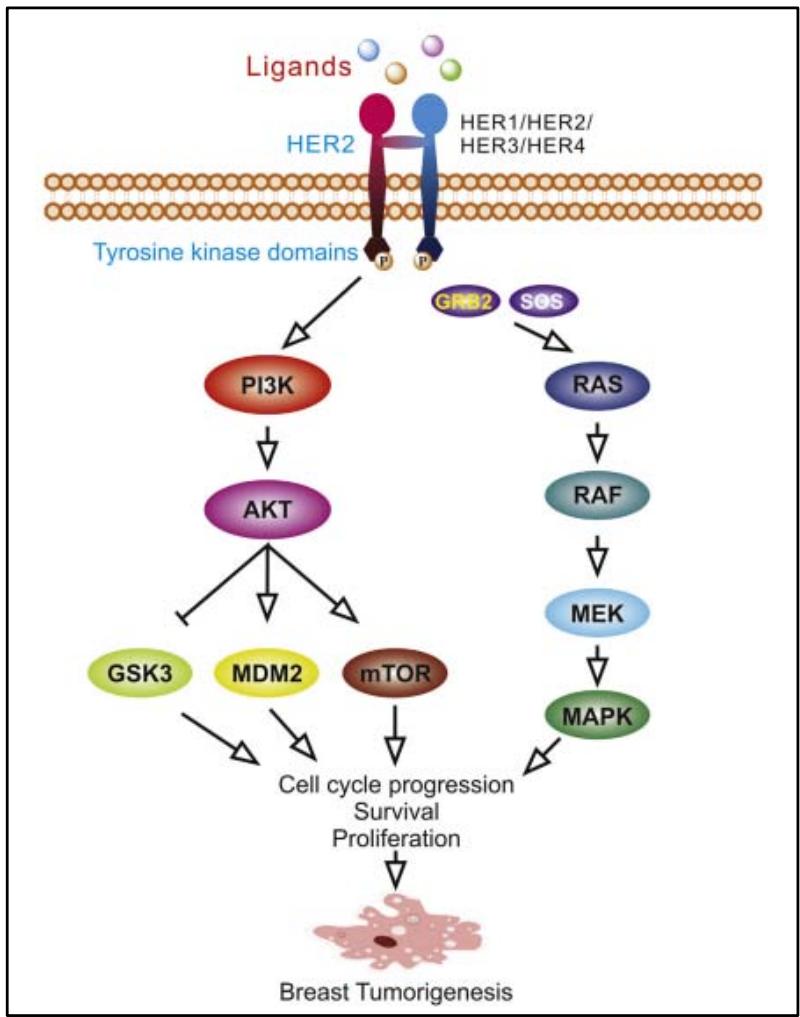 Figure 4: Phosphorylation of the tyrosine kinase domain of the HER2 receptor initiates downstream oncogenic signalling pathways, the main ones being the PI3K/AKT pathway and the Ras/MAPK pathway [1].
#### C/ Wnt/ 6 Catenin canonical signaling pathway
Wnt proteins are a family of highly glycosylated proteins that play an essential role in various developmental processes, including embryonic induction, generation of cell polarity as well as maintenance of adult tissue homeostasis. Canonical Wnt/β-catenin signaling is initiated by the binding of Wnt proteins to the two co-receptors Frizzled and Low-Density Lipoprotein Receptor-Related Protein 5 and 6 (LRP5/6). The Wnt/receptor interaction leads to the recruitment of Axin and Disheved proteins to the cell membrane, and induces inhibition of protein glycogen synthase kinase (GSK)-3b. The latter is a negative regulator of the Wnt pathway, which leads to the degradation of $\beta$ -catenin by the proteasome. Inhibition of GSK-3b leads to accumulation of $\beta$ -catenin in the cytoplasm and its subsequent translocation into the nucleus acts as a transcriptional Co activator in synergy with other transcription factors such as T-cell factor/lymphoid enhancing factor (TCF/LEF). $\beta$ -catenin regulates the transcription of several oncogenes, such as c-MYC, CCND1 and other target genes \[13\](Figure 5).
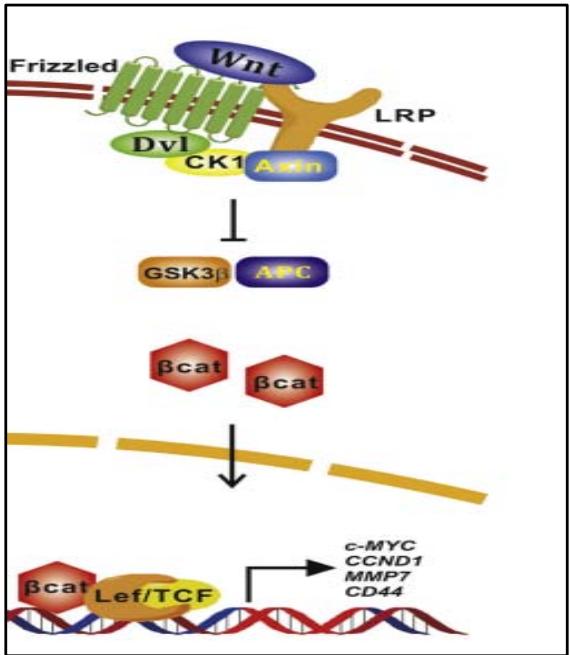 Figure 5: The Wnt ligand binds to frizzled membrane receptors and LRPs, leading to an accumulation of $\beta$ -catenin in the intracytoplasm. Once $\beta$ -catenin has been translocated to the nucleus, it activates transcription of oncogenes [1].
#### D/ Notch signalling pathway
Since the cloning of the Notch gene and identification of the structure of this receptor in the 1980s, several studies have been accumulating to confirm the role of this pathway in physiological processes, essentially neuronal differentiation, mesoderm induction during embryogenesis and the choice of commitment to B or T lymphoid lineages during hematopoiesis. It is reactivated in the carcinogenesis of solid tumors: bronchial cancer, breast cancer, pancreatic cancer, melanoma...and hematological malignancies [14].
The Notch pathway comprises four transmembrane receptors, Notch1 to Notch4 (N1-N4), and five ligands: Jagged1 and 2 (Jag1-2), and Delta-like1, 3 and 4 (DLL1, 3 and 4). The extracellular region of the receptor comprises a ligand-interacting domain consisting of 29 to 36 EGF-like motifs, plus 3 cysteine-rich LNR (Lin12 and Notch Repeats) motifs that prevent receptor activation irrespective of the presence or absence of ligand. The transmembrane region contains a heterodimerization domain (HD). The cytoplasmic portion includes a RAM domain for binding to its transcriptional partner (RBPJk), an ANK (Ankyrinrepeats) domain, two NLS nuclear localization signals, a TAD transactivation domain and a PEST domain on the C-terminal side, serving as a proteasomal degradation signal of the NICD \[14\](Figure 6).
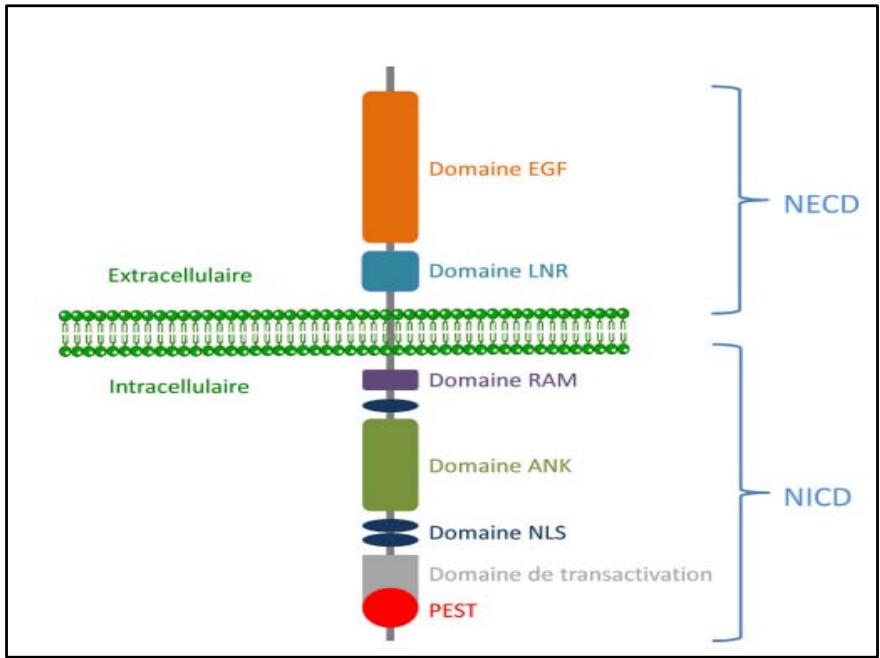 Figure 6: Schémas montrant les différents domaines constituant le récepteur de la voie NOTCH[14]
This signaling pathway is activated by the engagement of receptors with their ligands, expressed on a cell adjacent to the one receiving the signal. In the canonical pathway, ligand-receptor interaction leads to cleavage of the receptor, allowing release of its intracellular domain (NICD) and translocation to the nucleus. There, it associates with its transcriptional partner RBPJ $\kappa$, which is DNA-bound to the promoter of the pathway's target genes (Figure 7). In the absence of
NICD, RBPJ $\kappa$ is associated with transcriptional corepressors. Formation of the NICD-RBPJ $\kappa$ complex allows exclusion of these corepressors and recruitment of MAML (Master mind-like), which appears to serve as a scaffolding protein enabling formation of a transcriptional complex including other coactivators. This leads to chromatin opening and induction of transcription of target genes [15].
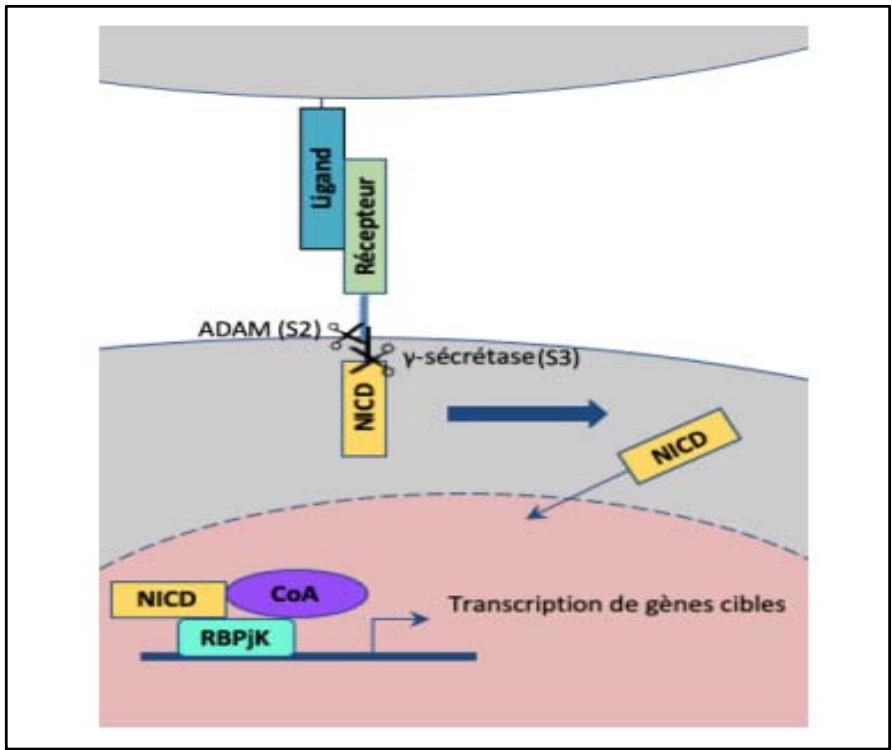 Figure 7: NOTCH signalling pathway: ligand-receptor interaction leads to cleavage of the Notch receptor, allowing release of its intracellular domain (NICD) and translocation to the nucleus. There it associates with its transcriptional partner RBPJk, which is bound to DNA to induce transcription of target genes [15].
Notch signaling has several direct target genes involved in cell cycle regulation. These include cyclins A, B and D1, and members of the Hes/Hey family. It also activates major oncogenic signaling pathways such as c-Myc, Ras and Wnt [16].
Notch signaling inhibits breast cancer cell apoptosis through various signaling pathways \[16\](Figure 8):
Activation of Akt signaling via NF $\kappa$ B, PI3K and mTOR signaling. Akt is responsible for direct inhibition of p53 or via the ASK1/JNK complex.
Activation of the c-Myc gene, which also has antiapoptotic activity.
Upregulation of survival by blocking apoptosis via direct and indirect inhibition of caspases.
Upregulates anti-apoptotic members, notably Bcl-2 and Bcl-XL, while downregulating pro-apoptotic members such as Bim and Noxa.
Notch signaling reduces the sensitivity of cells in triple-negative TNBC breast cancer to TRAIL death receptor-induced apoptosis.
Stimulation of the synthesis of cyclin-dependent kinase inhibitors p21 and p15, which also contribute to resistance to apoptosis.
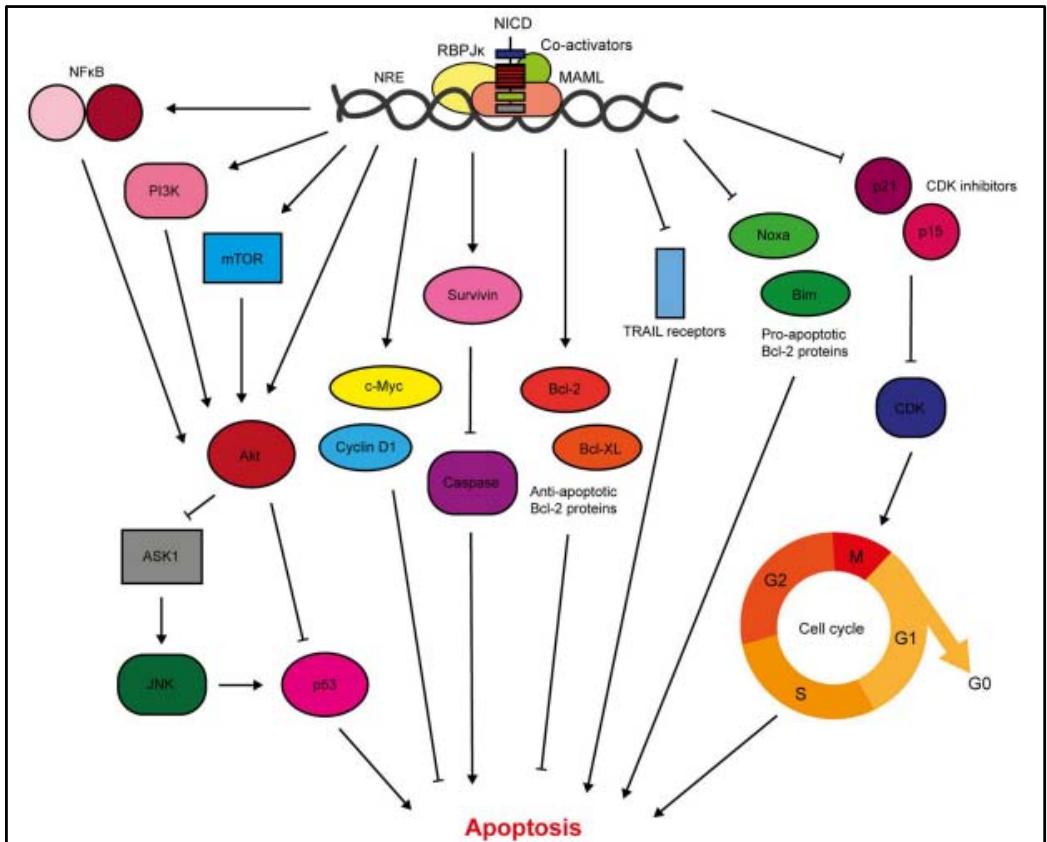 Figure 8: Diagram showing the role of the Notch pathway in regulating resistance to apoptosis [16].
Notch-mediated metastasis is induced primarily via TGF $\beta$ activation. It activates key regulators of the epithelial-mesenchymal transition EMT, notably the transcriptional repressors Slug and Snail, which mediate loss of cell-cell contacts through inhibition of E-cadherin expression. The mesenchymal markers ZEB1, $\beta$ -catenin, N-cadherin and vimentin are upregulated by Notch signaling. Also involved in invasion, it upregulates matrix-degrading enzymes including matrix metalloproteinases 2 and 9 and urokinase-type plasminogen activator (uPA), as well as $\beta 1$ -integrin [16].
Notch signaling is aberrantly activated in breast cancer. Overexpression of Notch receptors and ligands has been correlated with a poorer prognosis: resistance to chemotherapy and early recurrence. The data suggest that deregulation of Notch signaling is an early event in breast cancer tumorigenesis, with NICD accumulation in a wide range of subtypes, including ductal carcinoma in situ and epithelial hyperplasia. This implies that aberrant Notch signaling plays a causal role in breast tumor initiation [16].
Aberrant Notch activation may be secondary to mutations such as \[16\]:
Activating mutations in and around the PEST domain serving as a proteasomal degradation signal for the intracytoplasmic domain of the Notch1, 2 and 3 receptor;
Mutations disrupting the NLR, nuclear signaling motif, and heterodimerization domains;
Notch4 overexpression;
One cause of aberrant Notch signaling frequently found in breast cancer is the loss of the Numb protein. The Numb protein has long been known for its inhibitory role in the Notch signaling pathway. It opposes the Notch pathway by inhibiting recycling to the plasma membrane. It induces stabilization of the Notch ligand Delta-like 4 (Dll4) for degradation by lysosomes. NumB directly inhibits Notch by inducing polyubiquitination and preventing the activated intracytoplasmic domain from accessing the nucleus [17].
However, a recent Chinese study published in 2019 investigated the Notch pathway on nerve cells cultured in appropriate media, surprisingly showing that the NUMB protein enhances Notch signaling in a physiological way. In fact, the intracytoplasmic domain of the NOTCH type 1 receptor, N1ICD, undergoes various post-translational modifications, including ubiquitination by the BARD1-BRCA1 complex, facilitating degradation of the Notch receptor by the proteasome. The team discovered a new protein, BAP1, an enzyme capable of stabilizing N1ICD through its deubiquitination role and its ability to inhibit BRCA1. NUMB enhances Notch signalling by regulating the ubiquitin activity of the BAP1 protein and facilitating its association with N1ICD \[17\](Figure 9).
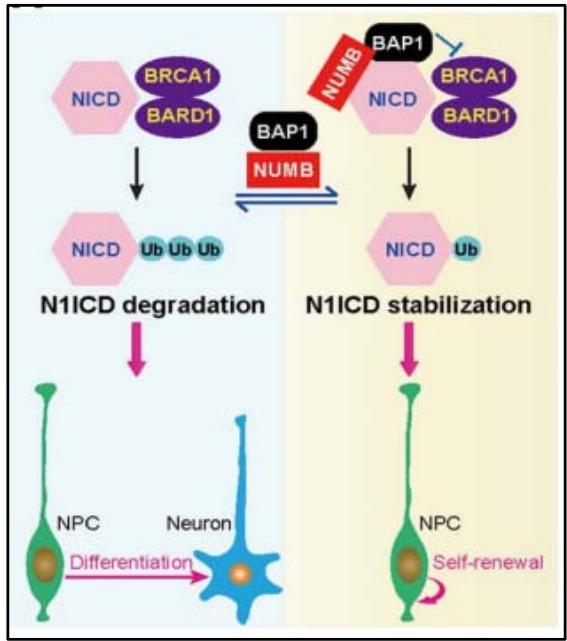 Figure 9: Schematic showing the role of NumB in stabilising N1ICD by associating with BAP1 to repress BRCA1/BARD1 ubiquitination activity in cortical neurons [17].
According to this new experiment, the importance long attributed to the Numb protein as a tumor suppressor in breast cancer is called into question. Explaining the relationship between loss of the Numb protein and breast cancer will be the subject of further exploration in the future.
#### E/ The sonichedgehog signaling pathway: SHH
The first identification of the sonichedgehog signalling pathway was made by Eric Wieschaus and Christiane Nüsslein-Volhard in 1980 during research on embryonic development in Drosophila melanogaster. This scientific paper won them the Nobel Prize for Physiology and Medicine in 1995.
Since its discovery, numerous studies have established the importance of SHH signaling in human embryogenesis and organogenesis, as well as its involvement in hematopoiesis. Normally, this pathway is inhibited in adults. However, it is reactivated in situations of tissue regeneration and stem cell renewal. Scientific research has identified three human homologues of the
Drosophila hedgehog gene: sonichedgehog, deserthededgehog and indianhedgehog, of which sonic hedgeorg is the best-studied ligand. This complexification is reflected in its receptor protein patch homolog (PTCH), of which there are two in humans: PTCH1 and PTCH2. However, PTCH1 remains the most widely expressed receptor in human cells.
Once the Sonic hedgehog (SHH) protein has been synthesized by the cell, it acts autocrine or paracrine on the target cells. The SHH factor then interacts with its receptor: the protein patch homolog, PTCH1, located on the primary cilium. The ligand-receptor interaction triggers internalization of this complex into endosomal vesicles. This internalization lifts the repression on a receptor called protein smoothened SMO, initially located in intracellular vesicles and repressed by PATCH1. SMO will travel to the primary cilium, where it will modulate the complex containing the Suppressor of Fused (SUFU) protein and the inactive form of a protein called Glioma-associated, GLI. Dissociation of the SUFU-GLI complex leads to degradation of the SUFU protein. In turn, the GLI factor undergoes transformations to acquire its active form. Activated GLI is translocated to the nucleus, where it specifically binds to sequences in the promoter regions of target genes, regulating their expression. These target genes include the GLI transcription factor itself, but also PTCH, cyclin D1 and products involved in the proliferation-differentiation balance (Figure 10). In the
presence of the SHH ligand, the PTCH1 receptor's repression of the SMO protein is lifted. SMO then acts on the SUFU/GLI complex. SUFU is degraded, while GLI is activated and translocated to the nucleus. It acts as a transcription factor for several target genes, namely: PTCH1, GLI1, FOXA2, BCL-2, BCL-XI, MYC and CYCLIN D1[18](Figure10).
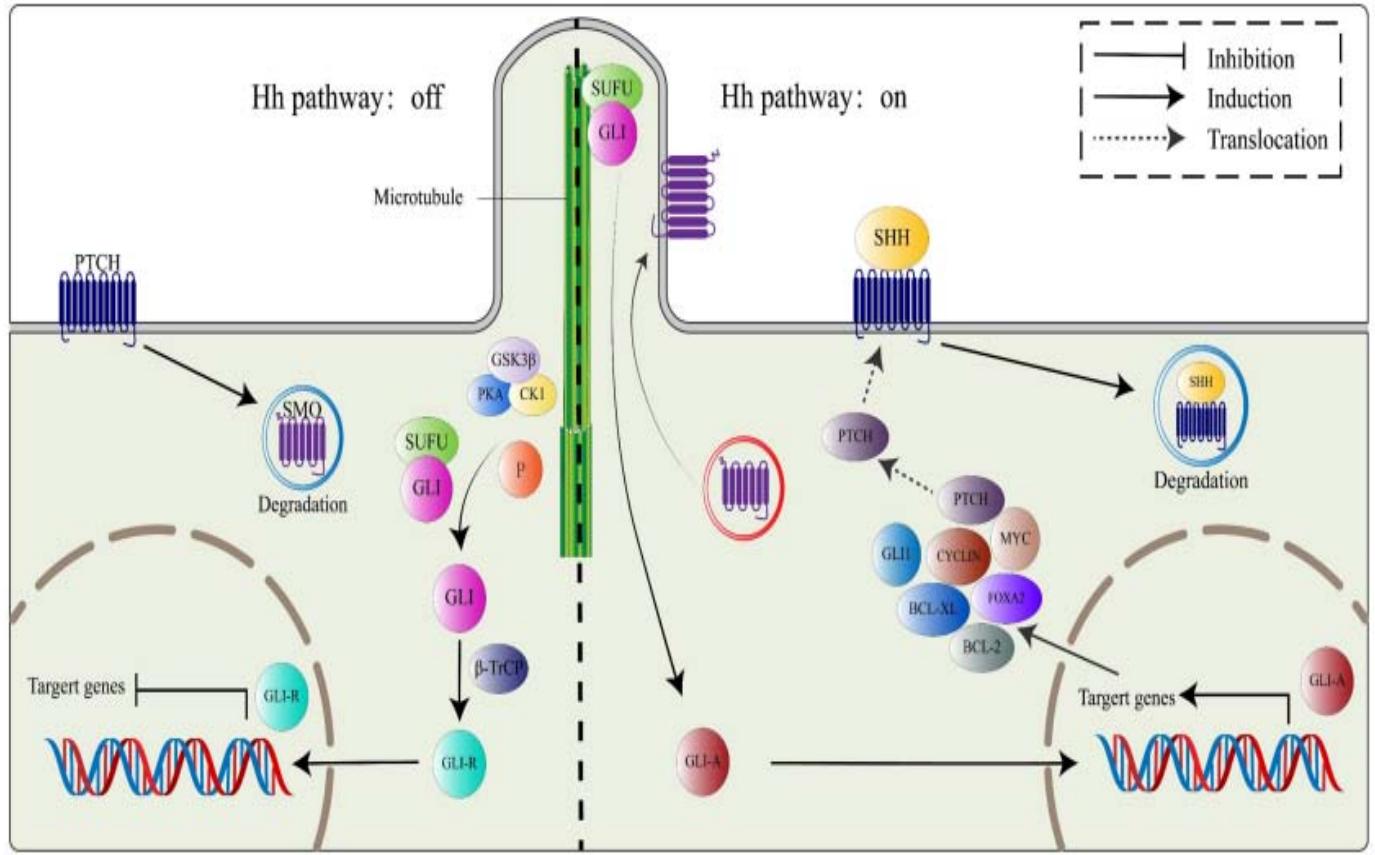 Figure 10: Diagram summarising the mechanism of action of the Sonic hedgehog SHH pathway: In the presence of the SHH ligand, the PTCH1 receptor lifts its repression of the SMO protein. SMO then acts on the SUFU/GLI complex. SUFU is degraded while GLI is activated and then translocated into the nucleus. IL acts as a transcription factor for several target genes, namely: PTCH1, GLI1, FOXA2, BCL-2, BCL-XI, MYC and CYCLIN D1[18].
Activation of the HH signaling pathway in breast cancer has been associated with presentation at a younger age, larger tumor size, presence of lymph node metastasis, negative progesterone receptor status, high proliferation index and poor overall survival [19].
Mutations in SHH, PTCH1 and GLI1 are very rare in breast cancer. The pathological involvement of the SHH pathway is explained by several epigenetic mechanisms, the most important of which are:
The transcription factor NF- $\kappa$ B (nuclear factor-kappa B) positively regulates SHH protein expression. It has been shown that an NF- $\kappa$ B-binding element is normally present in a CpG island of the SHH promoter. This site becomes accessible to NF- $\kappa$ B binding after demethylation. Reduced CpG methylation of the SHH promoter has been linked to increased SHH expression in several cancers, including breast cancer.
Low expression of $\text{PATCH1}$, which acts as a negative regulator of HH signalling. This correlates with the hyper-methylation of its promoter. However, SHH can bind with high affinity to receptors other than PTCH1, such as PTCH2, HHIP, complicating the interpretation of experimental results aimed at elucidating the involvement of PTCH in breast cancer.
High levels of GLI1 expression are more common in triple-negative and basal-like breast cancers. Experimental studies have also revealed a GLI1 mRNA splicing variant responsible for the shorter, truncated form of GLI1 (tGLI1). This variant is capable of increasing the expression of GLI-selective target genes, such as VEGF-A, CD24, MMP-2 and MMP-9, inducing more invasive and pro-angiogenic forms of breast cancer with very high metastatic potential [20].
F/ Cyclin D-dependent kinase signalling pathway
Cyclin D1 amplification is observed in almost $60\%$ of breast cancers. Estrogens also use cyclin D1 to exert their mitogenic effects. High cyclin D1 and HER2 overexpression have been reported to be associated with reduced recurrence-free survival and responsiveness to Tamoxifen [1].
G/ Mammary tumor kinase (BRK) signaling pathway
Also known as protein tyrosine kinase 6 (PTK6). It was originally cloned from a metastatic human breast tumor in 1994. The BRK transcript is encoded by an 8.93 kb DNA located on chromosome 20q13.3. The protein is a 451 amino acid kinase, comprising 3 parts, an SH3 domain and an SH2 domain, involved in protein- protein interactions, and a tyrosine kinase domain (SH1). Compared with members of the Src family, BRK lacks an amino-terminal myristoylation sequence (a mechanism used to position proteins precisely in specific membrane compartments). The modification consists in adding a fatty acid called myristate to one end of a protein) which makes the protein soluble and accessible for interactions with intracellular substrates [21].
Breast tumor kinase is overexpressed in $86\%$ of breast cancers [22]. The signaling events induced by PTK6 in the context of breast cancer are not well determined. However, it is clear that it is involved in several signaling pathways, summarized in figure 11.
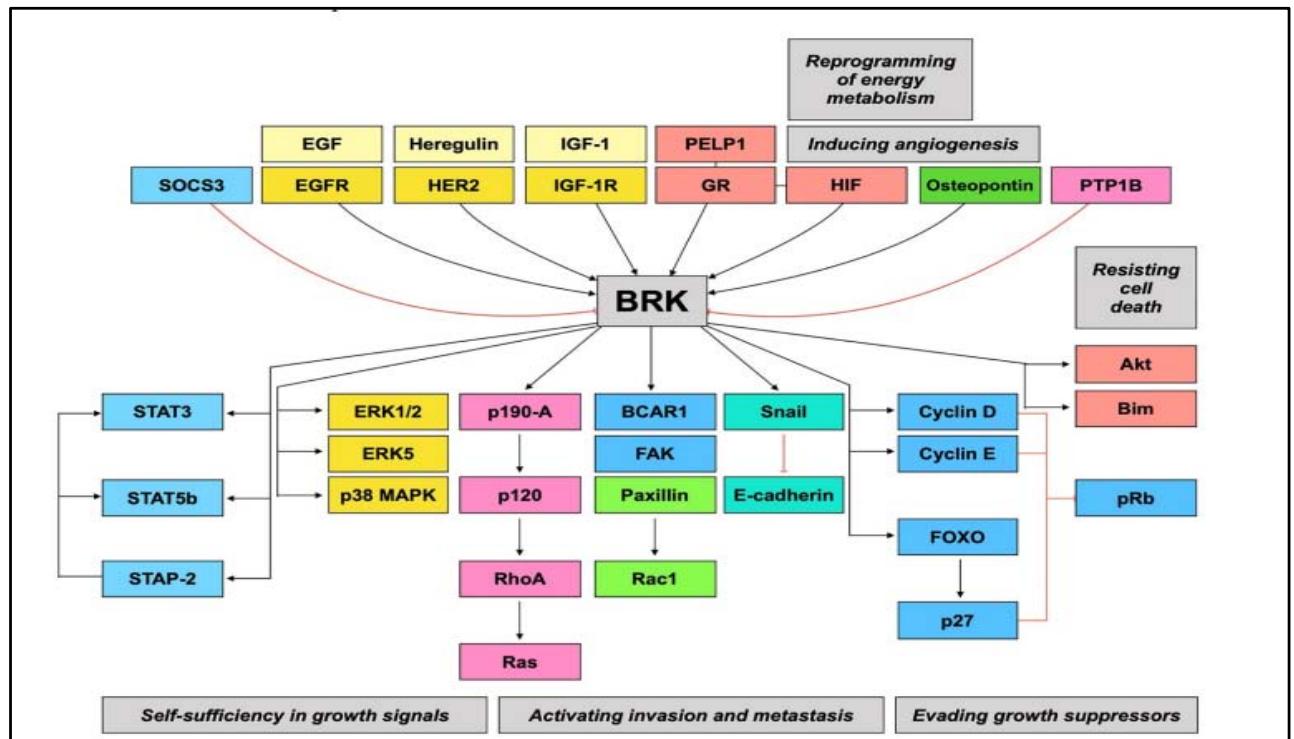 Figure 11: / The breast tumour kinase (BRK) signaling pathway is the convergence point for other signaling pathways that drive tumour progression [21].
H/ The PI3K/AKT/mTOR signaling pathway
Activation of the PI3K/AKT/mTOR signalling pathway is frequently found in breast cancer. The PI3PCA mutation is by far the most frequent mechanism, with mutation rates varying according to molecular subtype, with $49\%$ of mutations found within luminal A, $32\%$ within luminal B, $7\%$ within basal-like, and $42\%$ within HER2-enriched [23]. There are also numerous other mechanisms by which the PI3K signalling pathway is enhanced, such as HER2 amplification, IGF-1R overexpression, PTEN dysfunction and activating mutation of AKT1 [24].
Indeed, PI3P is made up of a catalytic subunit p110 and a regulatory subunit p85. There are three isoforms of p110, namely p110a (coded by PIK3CA), p110b and p110d. PI3K signalling is most often initiated either by the tyrosine kinase of the growth factor- activated receptor, or by the RAS protein, following a direct interaction with the p85 regulatory subunit, resulting in the recruitment of PI3K to the membrane [1]. PI3P assumes the phosphorylation of phosphatidylinositol-2-phosphate (PIP2) to phosphatidylinositol-3 phosphate (PIP3). This activating phosphorylation is finely regulated by the phosphatase PTEN (Phosphatase and tensinhomolog), whose role is to dephosphorylate PIP3 to PIP2. In this way, other downstream mediators are activated, AKT and mTOR leading to increased growth, translation, cell cycle progression and anti-apoptotic action [24].
The four main somatic mutations in PIK3CA are gain-of-function mutations involving four amino acids: E542K or E545K (glutamic acid at position 542/545 is replaced by lysine) located in exon 9, and H1047R (hestidine at position 1047 is replaced by arginine) or
H1047L (hestidine at position 1047 is replaced by leucine) located in exon 20. Mutations in exon 9 allow the p110α catalytic subunit to escape the inhibitory effect of p85. Mutations in exon 20 are located near the activation loop in the kinase domain. The mechanism by which they promote PI3K signaling is not well elucidated. [24].
Since its discovery in 2004, several studies have examined the prognostic and predictive value of PIK3CA gene mutations. One study showed that exon9 mutant patients were associated with a higher recurrence rate than exon20 mutant patients. [25]. Experimental and clinical evidence suggests that resistance to hormone therapy is largely due to hyperactivation of the phosphatidylinositol 3-kinase (PI3K) pathway. Breast cancers resistant to anti-estrogens often remain sensitive to hormone therapy combined with PI3K inhibitors [26] A 2018 analysis of 10329 patients with early-stage breast cancer found a significant association between early recurrence and PIK3CA mutations. In addition, PI3PCA mutations are predictive of poor response to anti-HER2 targeted therapy, with lower pCR (pathological complete response) rates than wild-type PI3PCA. [27].
## IV. HEREDITARY PREDISPOSITION TO BREAST CANCER
15-20% of breast cancers run in families: patients with breast cancer have one or more first- or second-degree relatives with the disease. High-risk genes, accounting for around 20% of familial risk, are BRCA1, BRCA2, TP53, STK11, CD1 and PTEN. It should be noted that over 50% of the genetic inheritance of familial breast cancer remains uncertain [28].
### A/ BRCA1/BRCA2 genes
In 1994, the BRCA1 gene was the first to be identified as a susceptibility gene for hereditary breast cancer. It is located on the long arm of chromosome 17 at 17q12-2. BRCA2 is located on chromosome 13, and was cloned in 1995[29]. Mutations in the BRCA1/BRCA2 genes are autosomal dominant. In the physiological state, BRCA proteins share a similar, cooperative tumor-suppressing mechanism by repairing DNA damage in double-strand breaks via homology-directed repair (HDR). Homologous recombination is based on faithful restoration of the damaged DNA sequence, using the homologous sequence of the undamaged chromosome as a template for repair. When this system is deficient, the relay is taken over by alternative DNA repair pathways that are much less genomically stable and considered mutagenic. This can lead to the activation of oncogenes or the inactivation of tumor suppressor genes, which explains the increased carcinogenic potential of these mutations [30]. However, homologous recombination primarily involves the detection of alterations by ATM (Ataxia telangiectasia mutated) and
ATR (Ataxia telangiectasia and RAD3-related) proteins and the mediation of signals by CHEK2 (checkpoint kinase 2) and BRCA1 itself. [29].
The Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) has made significant contributions to the characterization of the BRCA landscape. A recent update report on the CIMBA dataset summarized a total of 1650 and 1731 unique mutations in BRCA1 and BRCA2 respectively [31]. The most common type of mutation affecting these genes is the reading frame shift, leading mainly to the generation of premature stop codons and thus decreasing the levels of mature RNAs and functional proteins. [31].
Women with a BRCA1 mutation develop breast cancer with features similar to those found in the basal like non-hereditary subtype: young age (average 44 years), SBR grade III infiltrating ductal breast carcinoma (in $85\%$ of cases), extensive lymphocytic infiltrate, foci of necrosis, a very high proliferation index, Ki67, and a triple-negative phenotype. A p53 gene mutation is found in 50 to $77\%$ of cases, in contrast to sporadic forms, where it accounts for no more than $20\%$. Although BRCA1 mutant tumors are aggressive, they are more sensitive to cytotoxic agents, which improves their prognosis. In contrast, BRCA2 mutant tumors are heterogeneous, resembling sporadic tumors, with no dominant histological type, most often high-grade, and luminal in type. [29].
PARPs are enzymes involved in DNA repair for single-strand breaks. These repair pathways, via BRCA proteins (double-strand breaks) and PARP enzymes (single-strand breaks), are complementary: if one pathway is deficient and the other is blocked, the result is cell death by apoptosis, a phenomenon known as synthetic lethality. Single-strand breaks not repaired by PARP inhibition are converted into double-strand breaks during replication, unrepaired by the homologous repair system in the case of BRCA1 or BRCA2 mutation, leading to cell cycle arrest and apoptosis: this is known as double blockade (Figure 12). PARP inhibitors were first proposed and developed in ovarian cancer, then breast cancer, in cases of somatic or constitutional BRCA mutation, and more recently more widely, with significant positive results. The treatment received U.S. Food and Drug Administration (FDA) approval in the breast cancer indication in January 2018 [30].
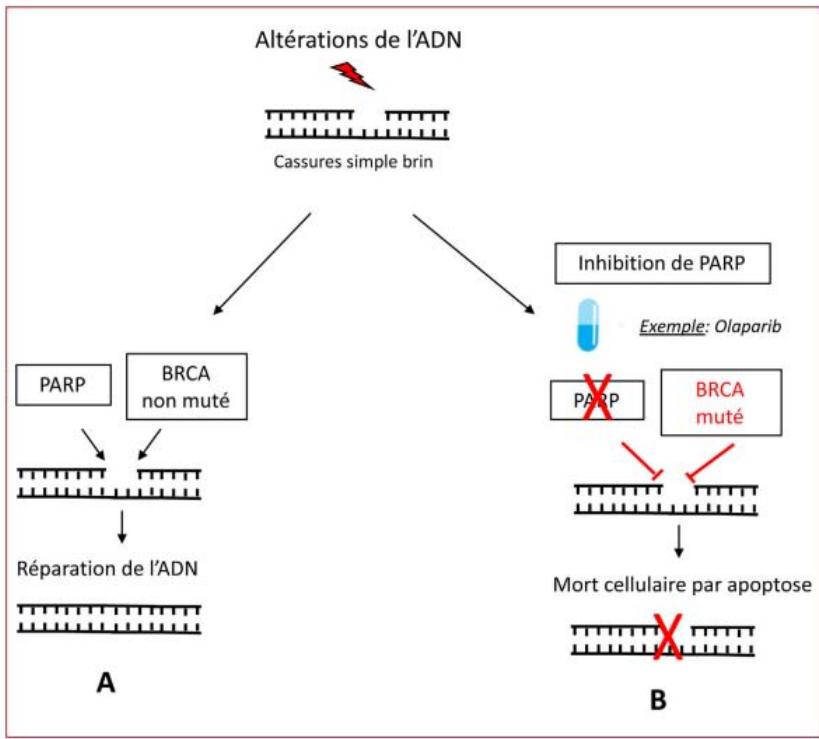 Figure 12: Schéma montrant le mode d'action des inhibiteurs de PARP: de lékalité synthétique [30]
#### B/ The PALB2 gene
The PALB2 gene was discovered in 2009. This gene is responsible for Fonconi anemia in the case of a biallelic mutation. Located on the lower short end of chromosome 16 (16p12.2), it codes for a protein enabling interaction between BRCA1, BRCA2 and other proteins involved in the repair of double-strand breaks. Indeed, the aminoterminal end of the PALB2 protein interacts with BRCA1 and RAD51, while its carboxyterminal end features a WD40 domain enabling interaction with BRCA2 and polymerases [32]. Recently, several studies have shown that patients with mutations in the PALB2 gene are just as likely to develop breast cancer as patients with mutations in the BRCA1 and BRA2 genes. These patients develop breast cancer of predominantly triple-negative phenotype, with a significantly shorter life expectancy than BRCA1-mutated patients. [29].
#### C/ The PTEN gene
The PTEN gene is a tumor suppressor gene located on chromosome 10, 10q23.3. It codes for a phosphatase that dephosphorylates phosphatidylinositol-3-phosphate (PIP3) to phosphatidylinositol-2-phosphate (PIP2), thus antagonizing the action of PI3K (phosphatidylinositol-3-kinase). A loss-of-function germline mutation in PTEN is responsible for a syndromic inherited predisposition: Cowden syndrome, an autosomal dominant disorder with variable age-related penetrance. This syndrome is manifested by damage to tissues derived from all three embryonic lineages [29]. Clinically, patients with this syndrome present with macrocephaly, mucocutaneous lesions (trichilemmomas, papulomatous papulles, etc.), benign lesions such as hamartomas, lipomas or fibroids [33]. In certain situations, clinical diagnosis is difficult, requiring molecular confirmation to retain the diagnosis [29].
50% of patients with this syndrome develop breast cancer by the age of 40. The histological appearance is characteristic: apocrine ductal carcinoma, most often with solid architecture surrounded by hyalinized collagen, with moderate to high mitotic activity and positive androgen receptors. [29][33].
#### D/ The TP53 gene
A germline mutation in the TP53 gene constitutes Li Fraumni syndrome. A second lesion affecting the second allele is required to initiate the process of carcinogenesis. Patients with this syndrome easily accumulate genetic abnormalities, and are at risk of developing cancers at a young age (< 30 years), or even during childhood. These patients may develop several cancers simultaneously. These include hard and soft tissue sarcomas (rhabdomyosarcomas), adrenocortical tumors, brain tumors and leukemias. [34]. Patients with this syndrome develop breast cancer in over $29\%$ of cases, without it being a characteristic histo-morphological feature. Malignant phyllodes tumours are also frequent. Male breast cancer is rarely described in the context of Li Fraumeni syndrome. [33]. Because of the major carcinogenic effects induced either by therapy: chemotherapy, radiotherapy or imaging-related irradiation, the latter present very restricted indications, hence the need to recognize this syndrome and confirm it by molecular study [33].
#### E/ The CDH1 gene
The CDH1 gene is located on the long arm of chromosome 16, 16q22.1. It codes for E-cadherin, a transmembrane intercellular adhesion protein involved in cell polarity and motility, as well as regulation of the microtubule network and organization of the actin cytoskeleton. A germline mutation in the CDH1 gene causes hereditary diffuse gastric cancer. Women with this mutation are at very high risk of developing lobular breast cancer, similar to that observed in women with BRCA1 or BRCA2 mutations. This cancer is observed either in the context of hereditary diffuse gastric cancer or without a family history of gastric cancer. Morphologically, lobular carcinoma developed in the context of a hereditary CDH1 mutation is similar to that observed in sporadic lobular carcinoma[29][33].
#### F/ The STK11 gene
Inactivation of the STK11 (Serine/Threonine Kinase 11) gene is responsible for Peutz-Jeghers syndrome, an autosomal dominant disease with complete penetrance. The incidence of this disease is estimated at one case per 100,000 births. This tumor suppressor gene is located on the short arm of chromosome 19, 19p13.3. It codes for a protein involved in the regulation of several cellular functions (such as signaling, apoptosis, cell polarity, regulation of the mTOR pathway), the most important of which is the limitation of cell proliferation in the event of restricted energy resources. Patients with Peutz-Jeghers syndrome clinically present with hyperpigmented macules on the oral mucosa, lips, palms and soles, as well as colonic and gastric hamartomatous polyps. These patients are at high risk of developing several types of cancer: colorectal, breast, small intestine, pancreatic, stomach and ovarian. In women with this mutation, the risk of developing breast cancer is around $15\%$, with a cumulative risk at age 70 of $45\%$.[29][34].
#### G/ The ATM gene
Germline mutation of the ATM gene, located on chromosome 11, is the cause of ataxia telangiectasia syndrome, an autosomal recessive disease with an estimated prevalence of 1 case per 40,000 to 100,000 births. These mutations can affect any region of the gene, and are of the following types: compound heterozygous nonsense mutations or a reading frame shift inducing deletions and insertions. This syndrome was first reported in 1957, and is characterized by progressive cerebellar ataxia revealed in early childhood, predominantly ocular telangiectasia, humoral immunodeficiency with low immunoglobulin production, and a predisposition to developing cancers, mainly lymphoid in children, as well as solid tumors, notably of
The breast and stomach. The degree of inactivity of the ATM protein means that the clinical presentation is highly heterogeneous, and benign forms with late-onset symptoms have also been described[35].
The ATM gene plays an important role in DNA double-strand break repair, apoptosis, cell cycle arrest and proliferation. It is also involved in the recombination of immunoglobulin chains and the TCR [34].
The risk of developing breast cancer in women with ataxia telangiectasia syndrome is very high, close to that of mutations affecting the BRCA genes. A recent study estimates that germline mutation of the ATM gene accounts for $1\%$ of all women followed for breast cancer. All molecular subtypes are associated with this mutation, with the exception of triple-negative breast cancer [35].
#### H/ The BRIP1 gene
The BRIP1 (BRCA1-interacting protein C-terminal helicase 1) gene, located on chromosome 17, 17q23, codes for a protein belonging to the helicase family. It interacts with numerous other proteins involved in regulating responses to double-strand DNA damage, notably BRCA1, as well as checkpoint signaling during DNA replication. In 2006, truncated heterozygous germline mutations were identified in a hereditary context of breast cancer without BRCA1 and BRCA2 gene abnormalities. The relative risk of developing breast cancer in heterozygous women is estimated at two. This type of mutation results in overexpression of a short protein unable to interact with the BRCA1 protein.[36]. Homozygous mutations are responsible for Fanconi anemia [37]. However, another oncogenic role has been suggested in addition to tumor suppression. The BRIP1 gene modulates the expression of several other genes involved in growth. Deletion of BRIP1 mRNA leads to cell cycle arrest in the G1/S phase and reduced expression of genes: cMYC, Ras GTPase, and the Rb gene [38]. Several recent studies have comparatively assessed BRIP1 protein and mRNA expression levels in various cultured breast cancer cell lines versus normal control breast cells. The results show significantly elevated expression levels of this protein in various molecular subtypes of breast cancer. However, out of 1651 genes studied, no mutations in the BRIP1 gene were found. This further demonstrates the dual behavior of the BRIP1 gene. The same study highlights the role of BRIP1 in tumor invasion and metastasis. High levels of BRIP1 mRNA expression are associated with decreased expression of metalloproteases, down-regulation of the MGAT5 gene, involved in cell growth and motility, and the chemokine CXCL12, the only ligand for CXCR4, involved in the formation of the pre-metastatic niche.[38][39]. The BRIP1 gene may be a potential molecular biomarker for predicting the prognosis of breast cancer patients that can replace conventional prognostic and analytical features such as lymph node status, tumor size, histological grade and molecular subtype [39].
Other genetic susceptibility genes for hereditary breast cancer include: NF1, NBN, CHEK2, RAD51C, RAD51D.
The various genes involved in hereditary breast cancer are currently the subject of genetic test kits (Oncotype DX and Mamatprint) whose published results are satisfactory for predicting the risk of breast cancer.
## V. CONCLUSION
The phenomenon of dedifferentiation is a recently described complex phenomenon that joins the two older theories, stochastic and hierarchical, in explaining the origin of the breast cancer cell. Indeed, the stochastic theory explains the variability of breast tumour subtypes by the diversity of oncogenic events undergone by stem and/or progenitor cells; whereas, according to the hierarchical theory, the same oncogenic event can generate several tumour cell lineages following the normal cellular hierarchy. Indeed, this subject is still a research question, in the sense that specifying the subtype and determining the cell of origin is a mandatory step in developing personalized treatments. The main signaling pathways involved in tumorigenesis: the estrogen receptor pathway, HER2, Wnt/βcatenin, are the same ones that regulate normal breast development and mammary stem cells. Given that only 20% of breast cancers occur in patients with a family history of the disease, we deduce that the genetic abnormalities found in breast cancer essentially involve lifestyle risk factors.
Links of interest: The authors declare that they have no links of interest.
Generating HTML Viewer...
References
39 Cites in Article
Y Feng (2018). Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis.
Nai Fu,Emma Nolan,Geoffrey Lindeman,Jane Visvader (2020). Stem Cells and the Differentiation Hierarchy in Mammary Gland Development.
Theresa Proia,Patricia Keller,Piyush Gupta,Ina Klebba,Ainsley Jones,Maja Sedic,Hannah Gilmore,Nadine Tung,Stephen Naber,Stuart Schnitt,Eric Lander,Charlotte Kuperwasser (2011). Genetic Predisposition Directs Breast Cancer Phenotype by Dictating Progenitor Cell Fate.
Luwei Tao,Dongxi Xiang,Ying Xie,Roderick Bronson,Zhe Li (2017). Induced p53 loss in mouse luminal cells causes clonal expansion and development of mammary tumours.
Chunyan Zhao,Karin Dahlman-Wright,Jan-Åke Gustafsson (2008). Estrogen Receptor β: An Overview and Update.
Antoni Hurtado,Kelly Holmes,Caryn Ross-Innes,Dominic Schmidt,Jason Carroll (2011). FOXA1 is a key determinant of estrogen receptor function and endocrine response.
Maria Pagano,Elena Ortona,Maria Dupuis (2020). A Role for Estrogen Receptor alpha36 in Cancer Progression.
N Fuentes,P Silveyra (2019). Estrogen receptor signaling mechanisms.
Peter Vrtačnik,Barbara Ostanek,Simona Mencej-Bedrač,Janja Marc (2014). The many faces of estrogen signaling.
Coralie Poulard,Isabelle Treilleux,Emilie Lavergne,Katia Bouchekioua‐bouzaghou,Sophie Goddard‐léon,Sylvie Chabaud,Olivier Trédan,Laura Corbo,Muriel Le Romancer (2012). Activation of rapid oestrogen signalling in aggressive human breast cancers.
Ilana Schlam,Sandra Swain (2021). HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now.
J.-C Xuhong,X.-W Qi,Y Zhang,J Jiang (2019). Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer.
Yang (2016). The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies.
Mehdi Brahmi,Olivia Bally,Lauriane Eberst,Philippe Cassier (2017). Ciblage thérapeutique de la voie Notch en oncologie.
M Ennajimi (2021). Rôle de la voie de signalisation Notch dans la différenciation des lymphocytes T CD8.
Abigail Edwards,Keith Brennan (2021). Notch Signalling in Breast Development and Cancer.
Z Luo (2020). NUMB enhances Notch signaling by repressing ubiquitination of NOTCH1 intracellular domain.
Chao Ma,Kang Hu,Irfan Ullah,Qing-Kang Zheng,Nan Zhang,Zhi-Gang Sun (2022). Molecular Mechanisms Involving the Sonic Hedgehog Pathway in Lung Cancer Therapy: Recent Advances.
Natalia Garcia,Mara Ulin,Ayman Al-Hendy,Qiwei Yang (2020). The Role of Hedgehog Pathway in Female Cancers.
Natalia Riobo-Del Galdo,Ángela Lara Montero,Eva Wertheimer (2019). Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics.
Hui Ang,Yi Yuan,Xianning Lai,Tuan Tan,Lingzhi Wang,Benjamin Huang,Vijay Pandey,Ruby Yun-Ju Huang,Peter Lobie,Boon Goh,Gautam Sethi,Celestial Yap,Ching Chan,Soo Lee,Alan Kumar (2021). Putting the BRK on breast cancer: From molecular target to therapeutics.
Amy Dwyer,Carlos Kerkvliet,Raisa Krutilina,Hilaire Playa,Deanna Parke,Warner Thomas,Branden Smeester,Branden Moriarity,Tiffany Seagroves,Carol Lange (2021). Breast Tumor Kinase (Brk/PTK6) Mediates Advanced Cancer Phenotypes via SH2-Domain Dependent Activation of RhoA and Aryl Hydrocarbon Receptor (AhR) Signaling.
(2012). Comprehensive molecular portraits of human breast tumours.
Toru Mukohara (2015). PI3K mutations in breast cancer: prognostic and therapeutic implications.
Sherene Loi,Stefan Michiels,Diether Lambrechts,Debora Fumagalli,Bart Claes,Pirkko-Liisa Kellokumpu-Lehtinen,Petri Bono,Vesa Kataja,Martine Piccart,Heikki Joensuu,Christos Sotiriou (2013). Somatic Mutation Profiling and Associations With Prognosis and Trastuzumab Benefit in Early Breast Cancer.
Todd Miller,Justin Balko,Carlos Arteaga (2011). Phosphatidylinositol 3-Kinase and Antiestrogen Resistance in Breast Cancer.
Hongwei Fan,Chao Li,Qian Xiang,Ling Xu,Zhuo Zhang,Qianxin Liu,Tonttong Zhang,Ying Zhou,Xia Zhao,Yimin Cui (2018). <scp><i>PIK3CA</i></scp> mutations and their response to neoadjuvant treatment in early breast cancer: <scp>A</scp> systematic review and meta‐analysis.
Camilla Wendt,Sara Margolin (2019). Identifying breast cancer susceptibility genes – a review of the genetic background in familial breast cancer.
Odile Cohen-Haguenauer (2019). Prédisposition héréditaire au cancer du sein (1).
M (2020). Syndrome héréditaire de prédisposition au cancer du sein et de l'ovaire : diagnostic et implications thérapeutiques.
A Piffer,E Luporsi,C Mathelin (2018). PALB2, gène majeur de susceptibilité au cancer du sein.
Anne Vincent-Salomon,Guillaume Bataillon,Lounes Djerroudi (2020). Les cancers héréditaires du sein vus par le pathologiste.
Florian Pesce,Mojgan Devouassoux-Shisheboran (2020). Les tumeurs héréditaires de l’ovaire vues par le pathologiste.
Luigia Stucci,Valeria Internò,Marco Tucci,Martina Perrone,Francesco Mannavola,Raffaele Palmirotta,Camillo Porta (2021). The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice.
Valeria Viassolo,Aurélie Ayme,Pierre Chappuis (2016). Cancer du sein : risque génétique.
Can-Bin Fang,Hua-Tao Wu,Man-Li Zhang,Jing Liu,Guo-Jun Zhang (2020). Fanconi Anemia Pathway: Mechanisms of Breast Cancer Predisposition Development and Potential Therapeutic Targets.
Balsam Rizeq,Saïd Sif,Gheyath Nasrallah,Allal Ouhtit (2020). Novel role of BRCA1 interacting C‐terminal helicase 1 ( <i>BRIP1)</i> in breast tumour cell invasion.
Umama Khan,Md. Khan (2021). Prognostic Value Estimation of BRIP1 in Breast Cancer by Exploiting Transcriptomics Data Through Bioinformatics Approaches.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Imane Eliahiai. 2026. \u201cOverview of the Main Signalling Pathways and Genetic Predispositions Involved in Breast Carcinogenesis.\u201d. Global Journal of Medical Research - C: Microbiology & Pathology GJMR-C Volume 24 (GJMR Volume 24 Issue C1): .
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Breast cancer remains the most deadly cancer in women worldwide. It is a highly heterogeneous disease group, both biologically and molecularly. Mammary carcinogenesis is a multi-stage, complex and progressive process, involving the accumulation of several genetic and epigenetic abnormalities in oncogenes and suppressor genes. These abnormalities lead to activation or inhibition of various molecules involved in cellular and molecular signaling pathways, thus altering stem cell proliferation, differentiation and cell death. Patients with certain constitutional genetic abnormalities are a sub-population at high risk of accumulating several molecular abnormalities at an early stage, and of developing more invasive breast cancers. Understanding the molecular pathogenesis of breast cancer is an essential step towards distinguishing molecular subtypes with different prognostic and therapeutic implications. This review provides a synthesis of the major molecular abnormalities found in breast cancer, focusing on molecules that are considered in the literature as prognostic or theranostic markers.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.