## I. LIPOSARCOMAS ARE ADIPOCYTIC SOFT TISSUE SARCOMAS
Soft tissue sarcomas (STS) are malignancies that show mesenchymal and neuroectodermal differentiation and thus most often resemble supportive and connective tissue including fat, blood vessels, muscle, bone, tendons, and nerves. Over 70 subtypes of sarcomas exist and pathologists have classified these broadly according to the degree to which they resemble differentiated cell types (Figure 1)[1]. This review will focus on the most common subset of STS in adults, "liposarcoma", which are tumors with histological features of specialized fat cells. Liposarcoma are broken down into several subtypes. The four with the highest incidence are: well- differentiated liposarcoma (WDLPS), dedifferentiated liposarcoma (DDLPS), myxoid liposarcoma (MLPS), and pleomorphic liposarcoma $(\mathsf{PLPS})^{1}$. Overall survival is highest for MLPS, followed by WDLPS and DDLPS, and then $\mathsf{PLPS}^{2-4}$ (Figure 2). While WDLPS occurs predominantly in the deep soft tissues of the limbs and retroperitoneum, DDLPS is located mostly in the retroperitoneum. MLPS and PLPS are preferentially located within the limbs[5]. Despite these broad categories, liposarcoma can also have mixed phenotypes and is often further subdivided into even more rare entities with other ultra-rare features. For instance, pleomorphic MLPS has attributes of both PLPS and $\mathsf{MLPS}^{6,7}$.
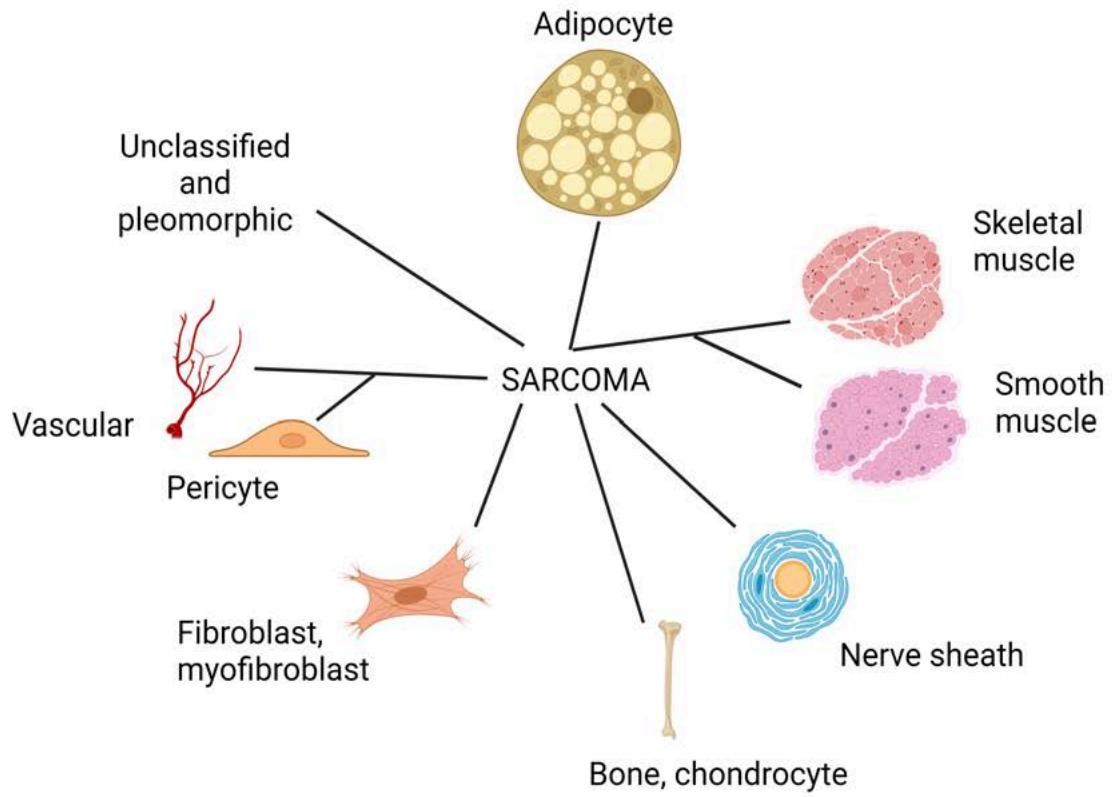 Figure 1: The Taxonomy of Soft Tissue Sarcoma. Sarcomas are classified according to pathologically defined tissue differentiation states. Liposarcomas are the adipocytic tumors
## II. LIPOSARCOMA GENOMIC CLASSIFICATIONS
STS have lower average somatic point mutation burdens than epithelial cancers. When examining their karyotypic characteristics, they are classically divided into two major groups: complex and simple. The liposarcoma subtypes WDLPS, DDLPS, and PLPS belong to the group of complex karyotypes, which are cells that have undergone steady and constant accumulation of multiple genomic copy number alterations, chromosomal anomalies and various types of rearrangements over time. This genomic instability is ongoing and occurs as a result of aberrations in genes involved in DNA repair, DNA replication and cell cycle regulation such as $TP53^{11}$. The complex karyotypes in these liposarcomas are likely to have arisen from mutations in the $TP53$ pathway. Both WDLPS and DDLPS have near universal amplification of chr12q, a region that includes MDM2, which is a gene that directs the protein degradation of $TP53$. For PLPS, recurrent mutations in $TP53$ (7%) and losses of RB1 occur[12,13].
Behavior and changes in the microenvironment can create a permissive context under which liposarcoma form. For instance, over expression of the immune-related cytokine IL-22 in a mouse on a high fat diet led to the only reported spontaneous formation of WDLPS in a mouse model $^{14}$. This implies that the relationship between the microenvironment and tumor may already be established when liposarcoma first form. This would explain why patient-derived WDLPS models have been difficult to establish as this dependence is still not well understood. Since chromosomal imbalances restrict the environment in which cancers can grow $^{15}$, the genomic instability that follows could then solidify this dependence.
Those sarcomas with simple karyotypes are nearly diploid; their driver events are typically fusion transcripts expressed via reciprocal chromosomal translocations. In the clinic, these diagnostic fusions are detected by fluorescent in situ hybridization (FISH), fusion panels, and reverse transcription polymerase chain reaction (RT-PCR). MLPS is an example of a liposarcoma with a simple karyotype and that is fusion-driven. It is mostly diploid and defined by a recurrent translocation between chromosomes 12 and 16: t(12;16) (q13;p11) that results in a fusion protein FUS-DDIT3.
## III. DEGREE OF ADIPOCYTIC DIFFERENTIATION ARE PATHOLOGIC MARKERS OF LIPOSARCOMA
Each liposarcoma subtype resembles different stages of adipocytic differentiation (Figure 2). This was first illustrated in an unsupervised analysis of gene expression patterns found in WDLPS, DDLPS, MLPS, PLPS, benign lipoma and normal fat $^{16}$. Three clusters formed: the first included normal fat, lipoma, and WDLPS; the second contained DDLPS and PLPS; and the third included only MLPS. In a complementary study, gene expression profiles of these four major liposarcoma subtypes were compared with those of human mesenchymal stem cells that were undergoing differentiation into mature fat. Each liposarcoma subtype resembled different stages in this process that were akin to their degree of differentiation $^{17}$. For instance, DDLPS expressed genes that were comparable to those that at day 7, which reflects stem cells in their early stages of differentiation, only starting their commitment to becoming fat as compared to cells at day 21, when maturation is almost complete. In support of this, 16 genes from the PPAR signaling pathway that leads to adipocytic terminal differentiation were significantly lower in DDLPS than in normal fat $^{18}$. On the contrary, WDLPS was more similar to cells at day 21, when differentiation is almost complete. PLPS closely resemble cells at day 10 while MLPS or round liposarcoma resembled those at day 14. These expression patterns imply that the degree of dedifferentiation of liposarcoma can be related to survival, with higher degree of differentiation leading to improved survival.
DNA methylation patterns also reflect these differences in differentiation states. When examining DNA methylation states in 80 various sarcomas in an unsupervised manner, each liposarcoma subtype formed a distinct group $^{19}$. Several distinguishing genes are related to adipocytic differentiation. One example is NNAT, which induces the activation of adipocytic transcription factors CREB and CEBP family $^{20}$ and was significantly methylated (hypermethylation) and upregulated in MLPS than in normal fat and other sarcomas $^{19,21}$. Decreased methylation (hypomethylation) and downregulation of NNAT was observed in DDLPS and PLPS, which likely results in a more dedifferentiated state. Another example is the CDKN2A gene, whose CpG island methylation levels are shared by PLPS, DDLPS, and non-neoplastic fat, but not MLPS $^{19}$. In addition, ALDH1A3 is involved in the oxidative degradation of lipids and may contribute to cancer stem potential. A strong negative correlation between the methylation of ALDH1A3 and its expression levels was found across several sarcoma subtypes, with the strongest hypermethylation and down regulation for MLPS $^{19}$.
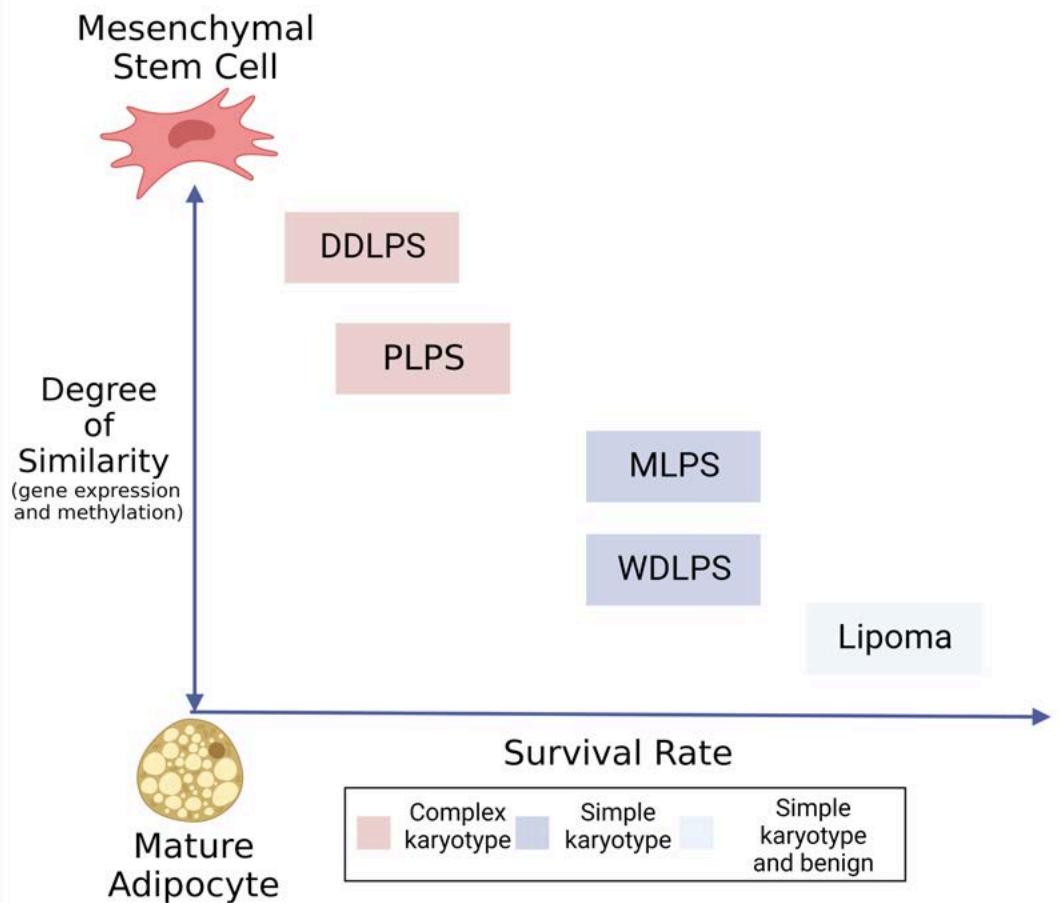 Figure 2: Survival and differentiation states of various liposarcoma subtypes
Dedifferentiated liposarcoma has the worst outcome, followed by pleomorphic, round-cell, then well-differentiated, and finally myxoid liposarcoma[22]. Gene expression and DNA methylation patterns in lipoma and liposarcoma subtypes are similar to those seen during the various stages along the differentiation pathway of mesenchymal stem cells as they progress towards becoming mature adipocytes. In concordance with these observations in liposarcoma, a recent hallmark of cancer – phenotypic plasticity – was recently added describing mechanisms associated with disrupted differentiation. The mechanisms are divided into three classes – dedifferentiation of mature cells to a progenitor or stem-like state, blocked differentiation preventing progenitor cells from maturing, and transdifferentiation enabling switching between lineages. It is likely that dedifferentiation and blocked differentiation occur in the various liposarcoma subtypes and that these two mechanisms are intertwined and held in place through mutations or epigenetic patterns.
## IV. LIPOSARCOMA FORMATION THROUGH GENETIC LOSS
Patients with Li-Fraumeni syndrome and retinoblastoma have germline mutations of TP53 and RB1, respectively, which leads to the formation of various tumor types, including high incidences of sarcomas as second and concurrent malignancies $^{23}$. In Li-Fraumeni patients, these include liposarcoma, which occur less frequently than other sarcoma subtypes such as osteosarcoma, leiomyosarcoma (LMS), and rhabdomyosarcoma $^{24,25}$. In like manner, retinoblastoma patients also occasionally develop liposarcoma, the majority of sarcoma risk being bone tumors, fibrosarcoma, rhabdomyosarcoma, and pleomorphic sarcomas $^{23}$. Therefore, there is evidence that these canonic cancer genes are responsible for driving liposarcoma initiation.
Sarcoma tumor initiation by mutation of TP53 and RB1 tumor suppressor genes have been demonstrated in vivo. Genetically engineered mouse or rat models of TP53 mutants develop sarcomas, namely angiosarcoma, osteosarcoma, and rhabdomyosarcoma with high levels of genomic instability $^{26-30}$. Deletion of both TP53 and RB1 genes in mice leads to lower tumor generation time, resulting in greatly reduced survival than is seen when each gene is mutated alone $^{31}$. When both TP53 and PTEN are simultaneously deleted specifically within adipose tissue, spontaneous generation of all four subtypes of liposarcoma occur $^{32}$. This model underscores the importance of the TP53 and PI3K/AKT pathways in the initiation of liposarcoma, which may be partly due to the way in which they activate the Notch signaling pathway $^{33}$. The effect of the
PI3K/AKT pathway on tumor initiation would also explain why PIK3CA amplification by fluorescent in situ hybridization (FISH) is associated with older age, larger tumor size, and shorter disease-free survival duration in liposarcoma, without distinction for a particular subtype $^{34}$. These models also illustrate how compounding gene losses can affect the nature and aggressiveness of the liposarcoma that is formed. This is further supported by the higher number of gene losses in DDLPS as compared to WDLPS $^{35}$.
TP53 and RB1 also alter the ability of mesenchymal cells to differentiate. Knocking out TP53 in mesenchymal stem cells (MSC) prevents the expression of PPAR $\gamma$, a key gene in directing adipocytic differentiation[36]. Instead, these MSC cells become more prone to osteogenic differentiation[36]. Without RB1, stem cells can no longer differentiate efficiently[37]. RB1 either pushes osteogenic differentiation in MSCs through RUNX2 or prevents adipocytic differentiation by inhibiting PPAR $\gamma$ [36,38].
Since Li-Fraumeni is an example of a syndrome with germline predisposition to developing multiple types of cancer including liposarcoma, there may be other germline risk factors to be identified. Out of 4,432 unique liposarcoma records in SEER (1973-2015 cohort), 2968 $(0.00063\%)$ had a recording of other concurrent cancers. Liposarcoma have concurrent diagnoses in ovarian cancer[39], hereditary nonpolyposis colorectal cancer[40], Muire-Torre syndrome[41], multiple myeloma[42] and CLL (also our recent unpublished data and infiltration in TCGA-SARC sample)[8]. Identifying these predisposition genes will enable us to interpret mutations in sporadic cases, as illustrated by the discovery of the telomere protection gene, POT1, as predisposing to angiosarcoma and cardiac sarcomas[39].
## V. TELOMERES IN LIPOSARCOMA SURVIVAL AND PERSISTENCE
Strategies to sustain cell survival include the elongation of chromosome ends: the telomeres. There are various Telomere Maintenance Mechanisms (TMM) including reactivation of the telomerase enzyme that serves to lengthen telomeres or the process of Alternative Lengthening of Telomeres (ALT) that employs homologous recombination methods to lengthen short telomeres. Activating mutations within the TERT promoter that encodes telomerase occur in a subset of MLPS $^{43}$. On the other hand, inactivating mutations and copy number losses in genes involved in ALT (ATRX DAXX) are detected in a subset of all liposarcomas, most frequently in DDLPS $^{44}$. Several assays are used to assess the activity of ALT within cells, which include: pulse field gel electrophoresis, terminal restriction fragment (TRF) Southern-blot analysis to measure telomere lengths, quantification of single-stranded circular DNA structures (C-circles) consisting of telomeric CCCTAA repeats, and immnofluorescence to identify the presence of ALT-associated promyelocytic leukemia bodies (APB). In all subtypes of liposarcoma, patients with ALT positivity as measured by these assays have worse progression-free and disease-specific survival rates $^{44-47}$. DDLPS is often the subtype cited with more ALT+ than WDLPS $^{48}$.
## VI. WELL-DIFFERENTIATED (WDLPS) AND DEDIFFERENTIATED LIPOSARCOMA (DDLPS)
Precursor or immature adipocytes are termed, "lipoblasts"49. Their gene expression patterns are most similar to those of nonmalignant adipocytes50. Well-differentiated (WDLPS) and dedifferentiated (DDLPS) liposarcomas are distinguishable from benign lipoblastoma and lipoma through karyotyping and breakpoint mapping. Lipoblastoma can have histological similarities with liposarcoma but is discriminated by an inversion involving the PLAG1 gene on chr851,52. Both lipoma and liposarcoma can have rearrangements or alterations on chr12. However, the breakpoints in lipoma appear to be more distal than in MLPS, WDLPS, and DDLPS53 with rearrangements involving HMGA2, rather than amplification of the entire gene. Therefore, the breakpoint location serves to identify disease type and severity within the adipose tissue. More recently, lipomas were shown to have low mutation burden, low copy number alterations (CNA) and share mutations with liposarcoma in APC, RYR2, and MAPK754.
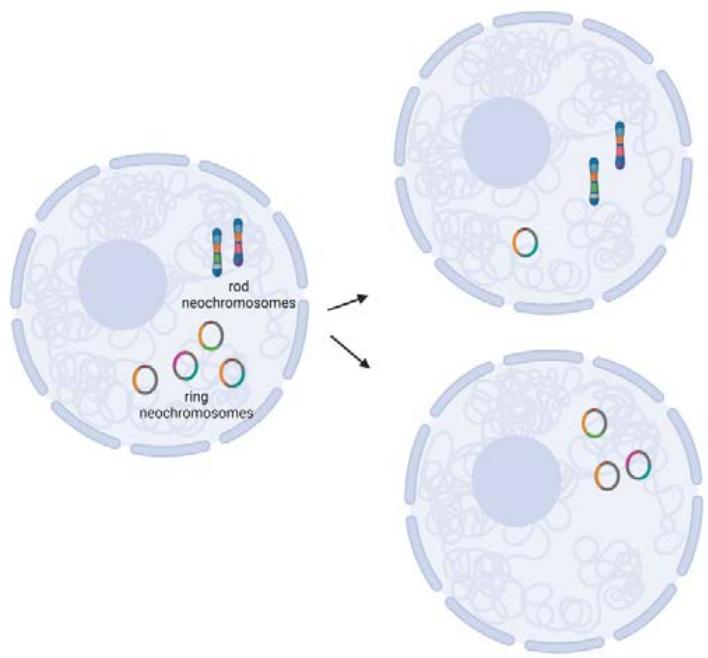
There is evidence that WDLPS and DDLPS share a common origin based on shared point mutations from which each subtype develops in an evolutionary divergence $^{35,55}$. There are patients who transition from WDLPS to DDLPS and very rarely, others who go from a diagnosis of DDLPS to WDLPS. In fact, each liposarcoma tumor is a mixture of both subtypes with one dominating over the other at different times. This common origin and plasticity are attributed to the presence of extraneous supernumerary ring or rod chromosomes within the nucleus, called "neochromosomes", amidst otherwise diploid-looking genomes. The neochromosomes are also common in atypical lipomatous tumors and have occasionally been reported in lipoblastoma $^{56-58}$. Whole genome sequencing of two isolated neochromosomes from a liposarcoma cell line revealed that they have no true centromeres and are therefore unstable $^{59}$. Upon closer molecular assessment using copy number microarrays and spectral karyotyping, these neochromosomes are derivations of chr12q13-15 along with other chromosomes, most commonly chr1q21-22 and chr6q23 $^{60-62}$. The observation of chr12 amplifications in both WDLPS and DDLPS is nearly universal $^{63}$. A minimum number of 20 copies per cell was observed using fluorescent in situ hybridization on the region that includes MDM2 and neighboring gene $CPM^{64}$.
Out of the four current theories on the formation of these neochromosomes$^{65}$, two have evidence that they are likely the primary source of genetic heterogeneity within liposarcoma tumors(Figure 5). The first is that chromosome shattering events, called, "chromothripsis", generated these neochromosomes. This suggests that this transformative event may have selected for cells with chr12 as their primary backbone, which promoted cell survival$^{59}$. This selection would seem most likely due to the most amplified genes: MDM2 and CDK4. MDM2 inhibits the tumor suppressor TP53, thereby circumventing the cell's rescue signals during DNA damage to repair without proceeding through the cell cycle(G1 and G2 arrest)and any signals towards apoptosis that would cause the aberrant cell to die. CDK4 would allow for unimpeded and enhanced progression through the cell cycle. The manner in which these chromosome pieces are stitched together into a neochromosome appear random. Therefore, just as no two snowflakes are alike, it is conceivable that the number and content of neochromosomes in each liposarcoma cell would not be the same and would change with each cell division in the same way that mitochondrial DNA populations are altered in each daughter cell. The second theory is based on whole genome data of two DDLPS specimens that did not exhibit any features of chromothripsis$^{1}$. In this study, the authors postulate that the neochromosomes are the result of progressive rearrangements and amplification. Both models are mutually exclusive and may delineate particular subsets of WDLPS and DDLPS. Following the generation of neochromosomes, either linear or circular breakage-fusion-bridge amplification (BFB) would lead to the multiple copies of the neochromosomes that are common to WDLPS and DDLPS $^{66}$. Since BFB events do not always cause the exact same breaks within a chromosome, the daughter cell of any given neochromosome-containing parental cell is likely to be different (Figure 5). This was demonstrated using a CRISPR-based ecTag method in glioblastoma spheroids $^{67}$. Amplification of oncogenes in extra chromosomal DNA may be the shortest route to heterogeneity than amplification of these genes within intact, autosomal chromosomes $^{67}$. Hence, there is vast heterogeneity within the population of liposarcoma cells, supporting the early observations that both WDLPS and DDLPS contain all four CD34/CD36 adipose markers by flow cytometry, with each of the four populations present at different proportions $^{68}$. The high level of heterogeneity is likely the reason treatment strategies are difficult to design. In addition, the triggers of transition or predominance of one subtype over the other is still unclear. Multiomic RNA and ATAC sequencing with spatial deconvolution may aid in tracking the mutation and environmental triggers as shown in recent studies in breast cancer and glioma $^{69}$.
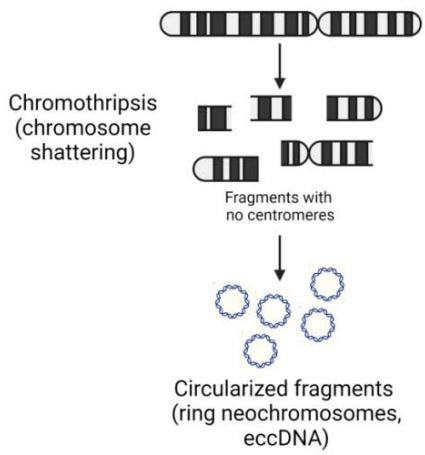 A
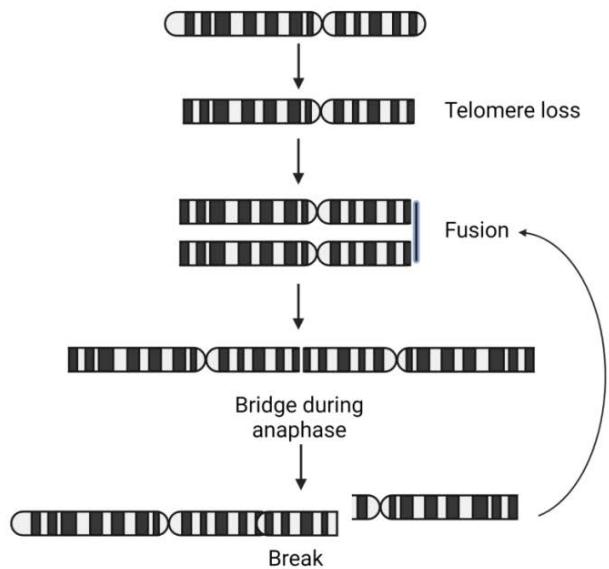 B Figure 3: Neochromosome formation in liposarcoma. A. Chromothripsis leads to chromosome fragments that then circularize into neochromosomes. B. The Break-Fusion-Bridge pathway that generate rod neochromosomes. These rods have the potential to circularize into ring neochromosomes
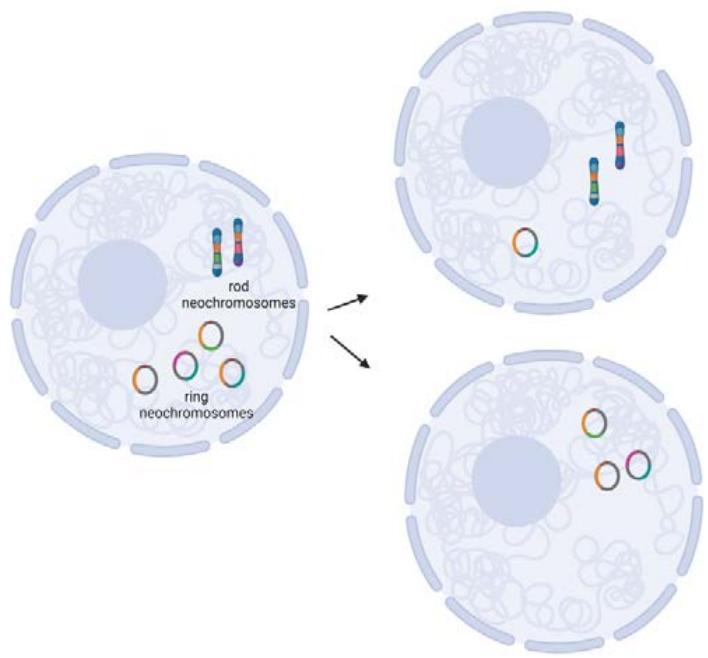 Figure 4: An illustration of the heterogeneity within an individual WDLPS or DDLPS due to the presence of neochromosomes. These nuclear neochromosomes are made up of different fragments from various chromosomes, chr12 being the most common and selected for (orange). The lack of true centromeres in these neochromosomes leads to the high probability of unequal segregation during mitosis, much like the random inheritance of mitochondrial DNA in daughter cells
Various genes within the region of chr12q amplification (MDM2, HMGA2, YEATS4, FRS2, CPM, DDIT3, PTPRQ) are implicated in the adipocytic differentiation pathways and in cancer progression. The degree to which each of these genes contribute to liposarcoma formation and progression is yet unclear. Evidence supporting roles for these genes is summarized below and in Tyler et al. $^{70}$.
MDM2: The N-terminal region promotes adipocyte differentiation through activation of CREB transcription at the expense of myogenesis in $P53^{- / - }$; $mdm2^{- / - }$ mouse embryonic fibroblasts $^{71}$. Mdm2 adipocytespecific knock-in (Mdm2-AKI) mice have increased white adipose tissue dysfunction, weight gain and insulin resistance when fed a high-fat diet $^{72}$.
CPM: CPM was significantly increased genes in early stages of differentiation when inducing adipogenesis in bone marrow derived human mesenchymal stem cells $^{73}$, adipose tissue-derived human mesenchymal stem cells $^{73}$, and adipose-derived stromal cells $^{74}$. Amplification that included CPM was observed in a large majority of WDLPS and DDLPS patient samples (78%, 39/50) $^{13}$. CPM distinguishes WDLPS and DDLPS from lipoma through having higher protein levels than benign lipoma and normal fat tissue $^{13}$. Knockdown using small interference RNA (siRNA) reduced cell proliferation, cell growth, colony formation, migration and invasion while increasing apoptosis in two of the DDLPS cell lines tested $^{13}$. This finding was recapitulated in eight liposarcoma cell lines that had undergone a genomewide CRISPER knockout screen (DepMap 22Q2 release)[75,76]. There, CPM was second most enriched dependency for viability among all the liposarcomalines.
DDIT3: DDIT3 (CHOP/GADD153) is a chromatin remodeler that is expressed highly during the last stages of adipocytic differentiation from lipoblasts to adipocytes $^{77}$. When over expressed in primitive sarcoma cells (fibrosarcoma) cells, DDIT3 can induce liposarcoma phenotypes $^{78}$. It is expressed at the protein level in WDLPS, DDLPS, MLPS, PLS, and lipoma $^{79}$. It blocks adipocytic differentiation by direct dominant negative inhibition of CEBP proteins from their target sites as well as preventing the accumulation of CEBPA in cells $^{80}$.
FRS2: FRS2 serves to recruit FGF, thereby facilitating FGFR signaling $^{81}$. FGFR signaling is also active during differentiation of mesenchymal stromal cells $^{82}$ and human pre-adipocytes $^{83,84}$. However, FRS2 inhibits adipocytic signaling in bone marrow stromal cells in 3D culture $^{85}$. These differing responses to FGFR signaling in cells according to environment and cell type that is receiving the signal may explain why not all liposarcoma have amplification of this gene.
HMGA2: FGF signaling also plays a role in HMGA2 expression. HMGA2 is a transcription factor that has relatively low expression in adult tissues as compared to embryonic and mesenchymal stem cells $^{86,87}$. Thus, it is important for proper development of multiple tissues and has high expression in the first three hours of adipogenesis of 3T3-L1 preadipocytes before decreasing in subsequent stages[88]. FGF signaling by adipocytic stem cells can induce HMGA2 expression[89]. Once turned on, it can bind Rb1 to displace HDAC1 from Rb/E2F at their binding sites, leading to activated E2F1 and cell cycle progression[90]. It is upregulated in lipomas and transgenic mice that overexpress HMGA2 result in hyperplasia of white adipose tissue[91,92]. These data suggest that HMGA2 alone cannot induce tumor progression and may only provide the proliferative context under which liposarcoma form.
PTPRQ: PTPRQ is a protein phosphatidylinositol phosphatase (PIPase) whose over expression would prevent adipocyte differentiation from mesenchymal stem cells $^{93}$. Gain in PTPRQ on chr12q21 was observed in $46\%$ of DDLPS $^{8}$.
YEATS4: By inhibiting the promoters of p14 and p21, YEATS4 (GAS4) represses the p53 pathway $^{94}$. Knockdown of YEATS4 in non-small cell lung cancer cells leads to increased expression of p21, p53 and PARP cleavage $^{95}$.
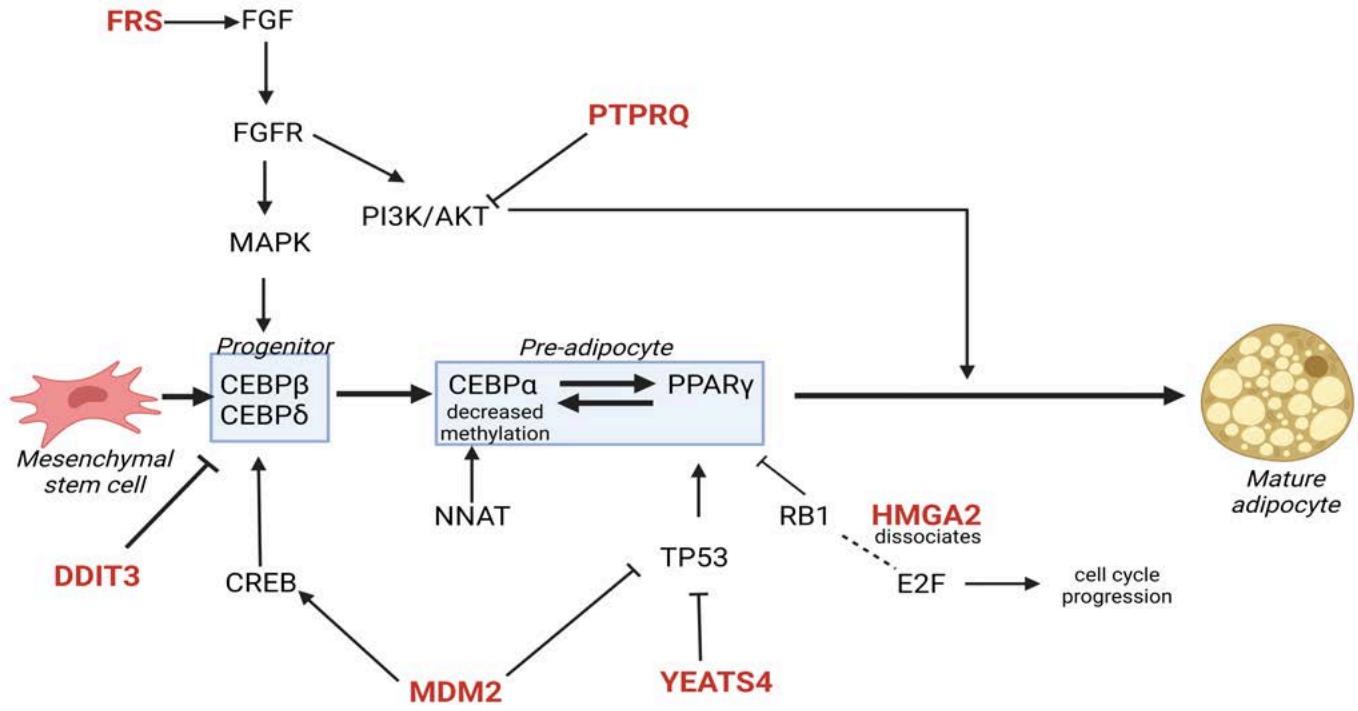 Figure 5: The role of genes from chr12 (in red) that are frequently amplified in WDLPS and DDLPS in adipocytic differentiation and tumor growth. The differentiation state of the tumor cell may affect the impact of these genes. Therefore, the selection of these genes for amplification may be determined by the cell differentiation state. FRS and DDIT3 have documented activities affecting the CEBP transcription factors that direct earlier adipocytic progenitors while YEATS4, HMGA2, and PTPRQ appear to affect the later pre-adipocytic stages
Besides chr12q, other copy number alterations involved in adipocyte differentiation are aberrant in WDLPS and DDLPS. Loss of methylation within the promoter of CEBPA (chr19q13) may explain the lower expression of CEBPA in DDLPS than in WDLPS $^{66}$. Gains in chr17p11 in DDLPS result in additional histologic features that are akin to $\mathrm{UPS}^{96}$. Gains in oncogenes that block adipocytic differentiation have been seen: JUN (chr1p32) $^{97}$ and YAP1 (chr11q22) $^{98,99}$. Lipid metabolism may be aberrant in DDLPS as losses and subsequent lower expression of genes such as PLIN2 (chr9p22), LIPE (chr19q13), DLAT (chr11q23-24), and ACAD8 (chr11q23-24) occur more often in DDLPS as compared to other sarcoma types $^{8,96}$. Rearrangement of SYT1 (calcium channel) was observed in WDLPS $^{100}$.
The level of heterogeneity and genomic complexity delineates differences between WDLPS and DDLPS. DDLPS have a higher number of point mutations that appear to be caused by the genome editing protein APOBEC (mutation signatures COSMIC2 and 13) $^{8}$. However, these point mutations may not contribute to the etiology of disease (passenger events) as the number was positively associated with age, largely nonfunctional, and not known to be cancer drivers $^{8,35}$. DDLPS also harbor higher number of rearrangements and copy number alterations than WLPS $^{35,101}$. In fact, the frequency of somatic copy number alterations was highest in DDLPS when compared against LMS, undifferentiated pleomorphic sarcoma (UPS), synovial sarcoma (SS), and malignant peripheral nerve sheath tumor (MPNST) $^{8}$. DDLPS has overall poorer survival, likely due to these increased burdens of mutations and copy number alterations $^{2-4}$. Further reduced local relapse-free survival was observed in DDLPS patients with loss of chr19q13 or chr9p22-24 or chr17q21 $^{96}$. When integrating both copy number and methylation alterations in a set of DDLPS (TCGA-SARC), disease-specific survival rate was significantly longer in one subgroup, cluster K3 (chr6q25.1 amplified and fewer unbalanced chromosome segments than K2) that shared a particular pattern of copy number alterations. Clusters K1 (JUN amplified) and K2 (TERT amplified and chromosomally unstable) had worse survival than $\mathrm{K}3^{8}$. This group had the lowest levels of immature dendritic cell infiltration. Overall, the study suggested that copy number alterations and methylation impacted survival and may be used as predictive biomarkers for DDLPS.
## VII. MYXOID LIPOSARCOMA (MLPS)
Myxoid liposarcoma are the most common liposarcoma in young patients under age $22^{6}$. The characteristic pathological features of myxoid liposarcoma are stellate spindle cells, signet-ring lipoblasts, "crow's feet" vascular network $^{102}$, and markers of immature adipogenicity $^{103}$. Transitional areas of increased cellularity can occur $^{104}$ with other patterns: round cell, pseudoacinar, lipoblast-rich, island, lipomatous, stromal hemangiopericytoma-like characteristics $^{105}$. The presence of small blue round cells in more than $5\%$ of the tumor is considered the "round-cell" subtype $^{104}$, which is more aggressive with poorer prognosis that metastasizes more frequently to the bone rather than to other tissue sites $^{104}$.
Within a background of a mostly quiet karyotype, the diagnostic molecular feature found in more than $95\%$ of tumors is the reciprocal translocation t(12;16) (q13;p11): FUS-DDIT3 (TLS-CHOP) $^{106}$. There are at least 10 known variants, of which the major categories involve breakpoints nearexon 5 or exon 7/8 of FUS, while other breakpoints occur after exon 4, 8, 13 in DDIT3 $^{107}$. These breakpoints eliminate the RNA-binding domain of FUS, which is then replaced by the DNA-binding domain of DDIT3 along with the rest of DDIT3 that includes a leucine zipper dimerization domain $^{108}$. Only variants with breakpoints near or after exon 13 have an intact RNA-binding domain from FUS in the fusion protein.
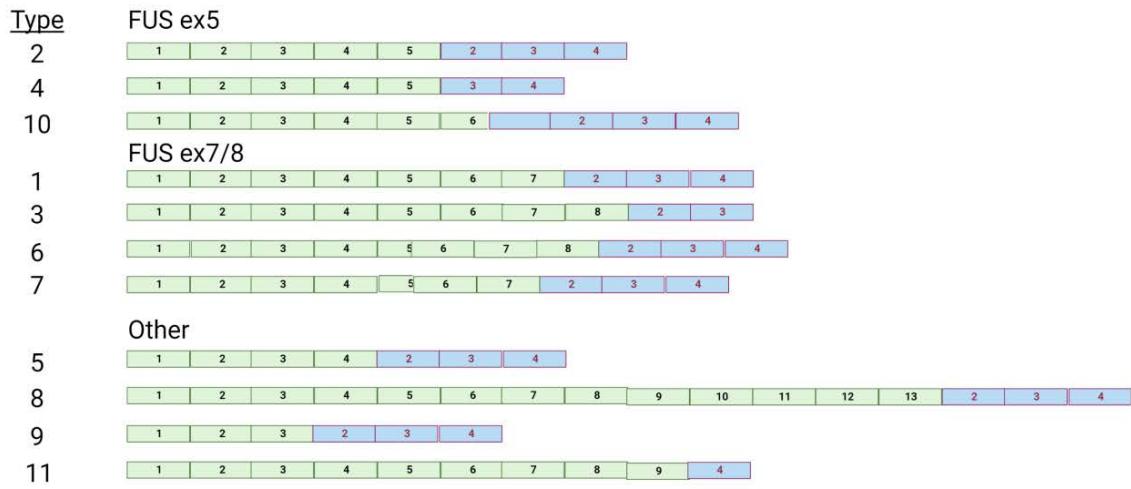 Figure 6: FUS-DDIT3 fusion transcript isoforms. FUS exons are shown in green, while DDIT3 exons are in blue
The FUS-DDIT3 fusion can transform mesenchymal cells in mice $^{109}$, partly by stimulating eIF4E expression, which results in down regulation of the PPAR $\gamma$ and C/EBP $\alpha$ pathways, thereby inhibiting adipocytic differentiation $^{110}$. It can also activate the IGFR1/PI3K/AKT pathway $^{110}$ and repress miR-486, which may result in upregulation of PAI-1, a molecular that is involved in tumor invasion and metastasis $^{111}$. An alternative mouse model demonstrated that expressing FUS-DDIT3 under a mesoderm promoter $Prx1$ within a p53 null background results in synergy in tumor formation $^{112}$. This may explain the poorer likelihood of survival in myxoid liposarcoma patients with TP53 mutations $^{113}$. Therefore, mutation in TP53 may contribute to a more aggressive tumor in the context of this translocation.
An alternative translocation event, EWSR1-DDIT3 (t(12;22)(q13;q12)), occurs in a minority of patients (4-5% in both pediatric and adult) (4-5%) with at least 4 known transcript isoforms $^{114}$. FUS and EWSR1 are functionally interchangeable since either gene fused to DDIT3 induced tumors in a xenograft model $^{115}$. In fact, FUS and EWSR1 are paralogs, belonging to the FET family of general RNA-binding proteins that also includes TAF15 $^{116}$. Together, these proteins appear to interact in a single complex $^{117,118}$ with a myriad of roles in RNA splicing, association with RNA helicases, DNA damage response, miRNA processing, RNA transport, and possibly others $^{116}$. Arginine methylation by the PRMT family, namely PRMT1, regulates their nucleocytoplasmic localization and binding to DNA $^{119}$.
Other distinguishing genomic features have been described for myxoid liposarcoma. The presence of the testis antigen NY-ESO-1 is thought to differentiate myxoid liposarcoma from other myxoid tumors $^{132}$. TERT promoter mutations are the most frequent in myxoid liposarcoma as compared to other sarcomas $^{43}$. Activating mutations in PIK3CA are the most common in myxoid liposarcoma as compared to other major liposarcoma histotypes $^{34}$, with greater incidence in round cell myxoid liposarcoma $^{133}$. These mutations appear to be mutually exclusive with PTEN loss and IGF1R expression $^{133}$. In addition, patients with PIK3CA mutations in the helical or kinase domains have a shorter disease-specific survival than those with wild type PIK3CA $^{134}$. Lower survival is also associated with methylation of the p14(ARF) promoter that leads to lower expression of ARF $^{135,136}$. Higher proliferative activity in MLPS is associated with high levels of $\beta$ -catenin $^{137}$ whereas growth through angiogenesis may be positively influenced by the hypermethylation and down regulation of the extracellular matrix glycoprotein EFEMP1, as compared to normal fat $^{19}$.
## VIII. PLEOMORPHIC LIPOSARCOMA (PLPS)
The definition of this particular subtype is the presence of pleomorphic lipoblasts $^{138}$. It is found frequently in the extremities of older adults, with those within upper extremities having poorer survival $^{139}$. This subtype excludes the distinguishing mutations found in the other subtypes described above: no fusions involving DDIT3 and no consistent amplification of $\mathsf{MDM2}^{140}$. It has a more complex karyotype than other liposarcoma subtypes $^{141,142}$, which may explain why these patients have the shortest survival of all liposarcoma subtypes. This complex karyotype nature of PLPS may form the basis for its pathologic and copy number profile resemblance to undifferentiated pleomorphic sarcoma (UPS) $^{138,143}$. Both had gains in: 1p, 1q, 5p, 19q, and 20q and recurrent losses in 1q, 2q, 3p, 4q, 10q, 11q, and 13q (including RB1). When comparing PLPS karyotypes among multiple complex karyotype pleomorphic sarcomas $^{144}$, the frequency of chromosomal aberrations was the fewest in pleomorphic liposarcoma. Thus, these may not be as advanced in complexity and severity as other pleomorphic sarcoma. Missense TP53 mutations within exons 5-9 were found in $60\%$ of the 31 cases that were examined $^{141,142}$, low levels of Rb1, and other features such as phyllodes of the breast $^{145}$ which occur more frequently in women with Li Fraumeni Syndrome (TP53 germline mutation).
## IX. CURRENT AND FUTURE GENOMICS
### a) Single Cell Sequencing
Despite the recent advances in understanding liposarcoma biology, there is still much to unravel in order to find effective targeted therapies for recurrent or metastatic lesions. Questions remain on how we can effectively explore themes within the complex heterogeneity of liposarcoma including degrees of adipocytic differentiation, mixed phenotype or clonal subtype, and cell of origin, which may enable avenues to potential therapeutics. Recently, single-cell sequencing (SCS) has made a dramatic impact on the field of cancer by revealing novel cell/differentiation states, exploring inter- and intra-tumor heterogeneity, and discovering rare cell populations previously undetected. Since Macosko et al. and Klein et al. developed Drop-Seq and in Drop respectively in 2015, approximately 14,534 articles were found using the keywords 'single-cell' and 'sequencing' to search in PubMed[146,147]. Among those articles, 68 contained the word 'sarcoma', and 5 contained 'liposarcoma'. This suggests that SCS is not being effectively used to explore sarcoma and liposarcoma genomics given the prevalence of SCS within the last decade. In the following section, we will discuss applying various SCS technologies to liposarcoma genomics, describe the common pitfalls when approaching liposarcomas, and examine the intersection of SCS and liposarcoma clinical care. Where liposarcoma-specific data are limited, we will extrapolate lessons learned from the cancer field and other sarcomas.
The democratization and commercialization of SCS have led to stable platforms for cancer research. The most widely used modality - transcriptomics or single-cell RNA-sequencing (scRNA-seq) -can profile gene expression for thousands of cells within a single experiment. The gene expression profile for each cell can be used to characterize and catalogue the cellular taxonomy of the tumor as well as define novel states or subtypes in cancer cells. Importantly, scRNA-seq has been used to detect rare subpopulations of cells including cancer stem cells and circulating tumor cells. For epigenomics, the most popular method is single-cell ATAC (assay for transposase-accessible chromatin) sequencing (scATAC-seq), which is used to measure the chromatin accessibility in single cells. Lastly, for genomics, single-cell DNA-sequencing (scDNA-seq) can be used for copy number alteration profiling, mutations, and clonal evolution. Additional layers of information can be studied through single-cell multiomics, where subsequent technologies can be used on cells of the same specimen followed by computational integration methods to combine the data or within the same cell where cellular barcodes link different -omics data.
While somatic hallmarks can be detected with techniques such as WES, SCS enables deeper exploration of mutations in the subpopulations within the tumor. Since both WDLPS and DDLPS contain amplifications within chr12q regions, SCS could be used to detect copy number alterations (CNAs) to separate malignant cells apart from normal cells and determine the clonal substructure of the malignant cells. This could enable understanding the cell of origin and how degrees of adipocytic could affect tumor burden. Technologies like Tapestri (Mission Bio) and Single-Cell CNV (10x Genomics) can directly detect CNAs by scDNA-seq. Recent work by the Navin group demonstrated that CNAs between scDNA-seq from single cells when merged together and bulk whole-exome-sequencing (WES) had high concordance for patients with triple negative breast cancer (TNBC). Pearson correlation showed a mean of 0.871 across five different matched patient data sets $^{148}$. A key limitation with this approach is that possible mutations for TNBC patients must be well-known in advance. This data feeds into a custom targeted panel of all known mutation sites for scDNA-seq, which greatly reduces the cost when compared to an unbiased panel. Advantages to using this approach, aside from the cost reduction, is enabling high-throughput single-cell analysis of clonal diversity within patients and understanding of possible clonal substructures. Key questions this could answer for WDLPS and DDLPS would be to understand the clonal evolution or transition, if it occurs, between WDLPS and DDLPS.
As an alternative to scDNA-seq, there are multiple software packages that can infer CNAs from scRNA-seq data. This has the advantage of utilizing the more popular scRNA-seq with the addition of evaluating gene expression $^{149,150}$. The currently available software packages used to infer CNAs from scRNA-seq data are: InferCNV, CaSpER, and CopyKAT $^{149,151,152}$. They operate under the assumption that CNAs are correlated with increasing or decreasing gene expression and that by fitting a mixture model to the data set, confounding factors from normal gene expression fluctuation could be removed. Work on synovial sarcoma, an aggressive neoplasm driven by the SS18-SSX fusion, demonstrated that CNAs could be detected using infer CNV on the scRNA-seq data and that the inferred CNAs matched the data from WES $^{153}$. The limitations with using software to infer CNAs from scRNA-seq is dependent on the model used with each software varying in detection of CNAs. In addition, normal reference cells may be required as an input and, in some cases, malignant and normal reference cells may not be easily distinguishable from the gene expression data alone. Nonetheless, these inference methods could determine tumor heterogeneity and enable identification of patient-specific features that are not found in the gene expression data.
Importantly for MLPS, which is driven by FUS-DDIT3, and in some cases, EWSR1-DDIT3 fusion, SCS can detect and quantitate fusions or structural rearrangements. However, this depends on the sequencing chemistry. There are four popular chemistries for generating sequencing reads - full-length, $3^{\prime}$, $5^{\prime}$, and tagmentation. $3^{\prime}$ and $5^{\prime}$ sequencing have been popularized by 10x Genomics, since these chemistries can easily enable profiling of up to 10,000 cells. However, these chemistries have high bias for $3^{\prime}$ or $5^{\prime}$ read coverage. This hinders the ability to detect mutations such as SNPs, indels, and rearrangements that may not exist at either $3^{\prime}$ or $5^{\prime}$ ends. In that regard, full-length mRNA profiling does enable in-depth sequencing capable of genotyping and detecting mutations. One such method that uses full-length mRNA sequencing is the SMART-seqwork flow (Takara Bio). However, SMART-seq has much lower throughput compared to $3^{\prime}$ and $5^{\prime}$ SCS. It requires fluorescent-activated cell sorting (FACS) to sort single-cells into wells of a 96-well plate. This does have an added benefit of cell typing the cells prior to sequencing if the cell type specific surface markers are well-expressed. Recently, SMART-seq was employed to detect the SS18-SSX fusion transcripts in synovial sarcoma $^{153}$. A common problem in SCS is annotating malignant cells v. normal cells. In this case, the presence of the fusion transcript was used to delineate malignant cells from normal cells. As for MLPS, since there are at least 10 known variants of the translocation, as known through synovial sarcoma, SMART-seq could easily identify the variants, while having the added benefit of transcriptomic data for each cell linked to any one variant. Importantly, regardless of grade, MLPS has potential to metastasize. A key question to explore using SCS would be to identify if there exists a cell state or subclone within the lesion that has a higher propensity for metastasis.
Feasibility of using SCS with fatty tissues, such as liposarcoma, is an important issue to resolve. Two major concerns with adipocytes are their large and fragile nature, which has proven to be a problem with SCS technologies. An alternative strategy to certain SCS methods, like scRNA-seq, which typically uses whole cells, is to use the nucleus - termed single-nucleus RNA-seq (snRNA-seq) $^{154}$. SnRNA-seq has previously been leveraged for various mouse and human adipose tissue $^{155-157}$. Recently, an atlas of white adipose tissue demonstrated that only snRNA-seq was capable of sequencing and detecting adipocytes, which were not present in the scRNA-seq data from the same tissue $^{158}$. Interestingly, while many of the other cells within the microenvironment were also present in snRNA-seq, there were also a lower abundance of endothelial and immune cells. Overall, this suggests that sequencing liposarcoma, where cases with WDLPS tend to be fattier, may require nucleus rather than whole cell. In that regard, techniques using scDNA-seq or scATAC-seq should not be affected since the nucleus is typically the default input.
### b) Cell-Free tumor DNA in Liposarcoma
Detection of possible recurrence or metastasis in patients with liposarcoma that have undergone complete resection can be difficult and costly. Because there are no diagnostic biomarkers associated with possible recurrence or metastasis, clinical examinations with frequent imaging throughout the body is the only alternative.
Recently, cell-free tumor DNA (ctDNA) has emerged as a novel method to interrogate cancer biology and etiology in a feasible manner by profiling tumor-derived materials, such as blood, cerebral spinal fluid, and urine $^{159}$. CtDNA often contain genetic material that had been shed from tumor cells, where such materials should reflect the tumor genome in some capacity. At a molecular level, somatic mutations, copy number alterations, methylation, and point mutations can be detected in ctDNA by sequencing methods. In that regard, ctDNA is a useful diagnostic tool that could detect early diagnosis and predict tumor burden and activity and overcome the hurdles of traditional diagnostic methods such as imaging and traditional biopsies.
Given that MLPS has a well-defined molecular diagnostic feature, the FUS-DDIT3 or the alternative EWSR1-DDIT3 translocation, ctDNA has recently been used to monitor disease activity of patients with MLPS $^{160}$. Quantification of ctDNA of the t(12;16) breakpoint for multiple patients using digital droplet PCR demonstrated a correlation of ctDNA concentration with tumor volume and stage. Upon resection there was an observed drop-off of t(12;16) ctDNA, while recurrence or metastases was associated with an increase of t(12;16) ctDNA concentration.
Interestingly, unlike MLPS where the translocation was detected by ctDNA, genotyping WDLPS and DDLPS was more difficult. While these tumors harbor amplifications resulting in multiple copies of MDM2, CDK4, and HMGA2, the method for detection by digital droplet PCR in a recent study was not nearly as sensitive $^{160}$. CtDNA derived from the peripheral blood from five WDLPS/DDLPS patients were collected and primers for MDM2 and a control gene, EIF2C1, were used to genotype. The MDM2/EIF2C1 ratio was 1.21 (range of 1.14-1.38), whereas health patients had a ratio of 1.09 (range of 0.69-1.41), which had no statistical significance, suggesting that PCR may not have enough specificity and sensitivity to detect the CNAs. On the other hand, a separate study used shallow whole-genome sequencing, which is well-established for genotyping with low-coverage, to detect MDM2 in ctDNA from the plasma of WDLPS and DDLPS patients $^{161}$. Interestingly, only two out of three DDLPS patients had readily detectable MDM2 amplification. This seemed to correlate with tumor size, where the undetected patient had a tumor size of $14~\mathrm{cm}$ v. 19 and $25~\mathrm{cm}$. Moreover, no WDLPS patients had detectable MDM2 amplification in ctDNA, perhaps due to the lower cellularity content of these tumors as opposed to DDLPS. In addition, a longitudinal study showed that MDM2 levels decreased after tumor resection. Overall, these data suggests that MDM2 amplification could be detected for DDLPS patients by shallow whole-genome sequencing from the plasma.
While PLPS is an aggressive sarcoma with high recurrences, it does not have a unifying genetic alteration that could be easily detected for disease monitoring. Over $50\%$ of patients diagnosed with PLPS will eventually have metastatic disease[22]. A study evaluating PLPS for biomarkers failed to identify prognostic biomarker for patients whose follow-up information was available $(n = 22)$ [12]. Despite the lack of distinctive genetic alterations, patient-specific gene variants found within the ctDNA could be a possible avenue for detecting residual disease or possible recurrence. One strategy would be to perform deep NGS sequencing on tumor tissue from surgical resection to discover patient specific alterations. Paired analysis of patient plasma from ctDNA using a targeted approach, like molecular tag-based sequencing, may reveal concordant mutations with the tumor tissue that could be used for disease monitoring during follow-up. In a recent study that monitored patient-specific ctDNA across a diverse set of tumors, the authors found that for patients $(n = 40)$ with three or more longitudinal time points the patient-specific ctDNA had a correlation with tumor burden in 16/19 (85%) patients with partial response and overall in 27/40 (68%) patients $^{162}$. On the other hand, use of cancer antigens only correlated with tumor burden in 19/40 (47.5%) patients, suggesting a lower utility than patient-specific ctDNA.
Outside of somatic mutations, detection of DNA methylation in ctDNA may offer an alternative modality for monitoring tumor burden and recurrence. Whole-genome bisulfite sequencing (WGBS) can detect DNA methylation throughout the genome. Importantly, methylation patterns greatly differ between malignant and normal cells, and could be used to distinguish between different cancer types. Certain sarcomas, such as synovial sarcomas, had unique methylation patterns that was relatively uniform. On the other hand, DDLPS had 3-4 methylation patterns that overlapped with undifferentiated pleomorphic sarcoma and gynecologic leiomyosarcoma. Nonetheless, detecting methylation in ctDNA has utility for monitoring tumor burden. The Circulating Cell-free Genome Atlas (CCGA; NCT02889978) is a prospective, multi-center, observational study that uses machine learning to detect cancer type and tumor burden from ctDNA $^{163}$. By WGBS, methylation signatures could robustly identify several cancer types with high specificity. Importantly, they found that WGBS of ctDNA outperformed WGS, which detected somatic mutations, and targeted mutation panels in classifying cancer types. Because methylation is more pervasive than mutations, it may enable lower limits of detection compared to detection limits for somatic mutations detected through WGS or targeted ctDNA panels $^{164}$. A clear limitation in this study is the small number of sarcoma patients included. Another limitation is that not all participants were asymptomatic, could inform the utility of DNA methylation for disease monitoring. Studies including asymptomatic patients were still ongoing.
## X. SUMMARY
In summary, WDLPS, DDLPS and PLPS have complex genomics due to either formation or propagation of neochromosomes or complex rearrangements and copy number alterations. These mutations lead to high levels of heterogeneity generating mixed tumor phenotypes, which can be difficult to classify. The altered genes, which are selected for during tumor evolution, drive the perpetual survival and continued growth of immature or poorly differentiated dipocytes. Unlike the other liposarcoma subtypes, MLPS is characterized by a translocation, where the N-terminal partner, DDIT3, in a healthy context plays an important role in regulating adipogenic differentiation. However, in the setting of MLPS, the
The latest developments in tools and technologies, including SCS and ctDNA, will be fundamental in advancing biology, diagnostics, and molecular therapeutics. SCS may shed light on intertumoral heterogeneity and identify subclones with actionable gene targets. Utilizing ctDNA may enable a feasible method for diagnosis and disease monitoring where recurrence is a possibility. Most importantly, continued exploration of the genomics of liposarcoma should enable advances in drug development centered on the genetic alterations.
### ACKNOWLEDGEMENTS
To Jing Zheng for SEER investigation. To Andrew Ibrahim, and Enes Kelestemur for reviews. All original figures were generated in BioRender.com.
Generating HTML Viewer...
References
151 Cites in Article
Barry Taylor,Jordi Barretina,Robert Maki,Cristina Antonescu,Samuel Singer,Marc Ladanyi (2011). Advances in sarcoma genomics and new therapeutic targets.
Eric Stanelle,Emily Christison-Lagay,Emma Sidebotham,Samuel Singer,Cristina Antonescu,Paul Meyers,Michael La Quaglia (2012). Prognostic Factors and Survival in Pediatric and Adolescent Liposarcoma.
Yoon Oh,Seong Yi,Ki Kim,Yong Cho,Seung Beum,Young Lee,Jin-Suck Suh,Hyuk Hur,Kyung Kim,Sung Kim,Young Choi,Kyoo-Ho Shin,Hyun Jun,Sung Kim,Jeeyun Lee,Se Park,Sung Noh,Sun Rha,Hyo Kim (2016). Prognostic Model to Predict Survival Outcome for Curatively Resected Liposarcoma: A Multi-Institutional Experience.
Carolin Knebel,Ulrich Lenze,Florian Pohlig,Florian Lenze,Norbert Harrasser,Christian Suren,Jonathan Breitenbach,Hans Rechl,Rüdiger Von Eisenhart-Rothe,Heinrich Mühlhofer (2017). Prognostic factors and outcome of Liposarcoma patients: a retrospective evaluation over 15 years.
S Schwartz,S Centurion (2019). Unknown Title.
Rita Alaggio,Cheryl Coffin,Sharon Weiss,Julia Bridge,Josephine Issakov,Andre Oliveira,Andrew Folpe (2009). Liposarcomas in Young Patients.
Tiffany Sinclair,Chad Thorson,Elysia Alvarez,Serena Tan,Sheri Spunt,Stephanie Chao (2017). Pleomorphic myxoid liposarcoma in an adolescent with Li–Fraumeni syndrome.
Zheng Wang,Ling Yang,Yuhui Jiang,Zhi-Qiang Ling,Zhigang Li,Yuan Cheng,Heng Huang,Lingdi Wang,Yi Pan,Zhenzhen Wang,Xiaoqiang Yan,Yan Chen (2011). High Fat Diet Induces Formation of Spontaneous Liposarcoma in Mouse Adipose Tissue with Overexpression of Interleukin 22.
Uri Ben-David,Angelika Amon (2020). Context is everything: aneuploidy in cancer.
Samuel Singer,Nicholas Socci,Grazia Ambrosini,Elliot Sambol,Penelope Decarolis,Yuhsin Wu,Rachael O'connor,Robert Maki,Agnes Viale,Chris Sander,Gary Schwartz,Cristina Antonescu (2007). Gene Expression Profiling of Liposarcoma Identifies Distinct Biological Types/Subtypes and Potential Therapeutic Targets in Well-Differentiated and Dedifferentiated Liposarcoma.
Igor Matushansky,Eva Hernando,Nicholas Socci,Tulio Matos,Joslyn Mills,Mark Edgar,Gary Schwartz,Samuel Singer,Carlos Cordon-Cardo,Robert Maki (2008). A Developmental Model of Sarcomagenesis Defines a Differentiation-Based Classification for Liposarcomas.
Hongliang Yu,Dong Pei,Longyun Chen,Xiaoxiang Zhou,Haiwen Zhu (2017). Identification of key genes and molecular mechanisms associated with dedifferentiated liposarcoma based on bioinformatic methods.
Marcus Renner,Thomas Wolf,Hannah Meyer,Wolfgang Hartmann,Roland Penzel,Alexis Ulrich,Burkhard Lehner,Volker Hovestadt,Esteban Czwan,Gerlinde Egerer,Thomas Schmitt,Ingo Alldinger,Eva Renker,Volker Ehemann,Roland Eils,Eva Wardelmann,Reinhard Büttner,Peter Lichter,Benedikt Brors,Peter Schirmacher,Gunhild Mechtersheimer (2013). Integrative DNA methylation and gene expression analysis in high-grade soft tissue sarcomas.
Young Suh,Won Kim,Changsuk Moon,Yun Hong,Su-Yong Eun,Joo Lim,Joo Choi,Jihyun Song,Myeong Jung (2005). Ectopic expression of Neuronatin potentiates adipogenesis through enhanced phosphorylation of cAMP-response element-binding protein in 3T3-L1 cells.
Sofia Thelin-J�rnum,Carin Lassen,Ioannis Panagopoulos,Nils Mandahl,Pierre �man (1999). Identification of genes differentially expressed in TLS-CHOP carrying myxoid liposarcomas.
Kimberly Dalal,Michael Kattan,Cristina Antonescu,Murray Brennan,Samuel Singer (2006). Subtype Specific Prognostic Nomogram for Patients With Primary Liposarcoma of the Retroperitoneum, Extremity, or Trunk.
Ruth Kleinerman,Sara Schonfeld,Margaret Tucker (2012). Sarcomas in hereditary retinoblastoma.
F Li (1988). A cancer family syndrome in twentyfour kindreds.
P Kleihues,B Schauble,A Zur Hausen,J Esteve,H Ohgaki (1997). Tumors associated with p53 germline mutations: a synopsis of 91 families.
G Liu (2004). Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice.
Michele Harvey,Hannes Vogel,Danna Morris,Allan Bradley,Alan Bernstein,Lawrence Donehower (1995). A mutant p53 transgene accelerates tumour development in heterozygous but not nullizygous p53–deficient mice.
Aaron Mccoy,Cynthia Besch-Williford,Craig Franklin,Edward Weinstein,Xiaoxia Cui (2013). Creation and preliminary characterization of a <i>Tp53</i> knockout rat.
Roel Hermsen,Pim Toonen,Ewart Kuijk,Sameh Youssef,Raoul Kuiper,Sebastiaan Van Heesch,Alain De Bruin,Edwin Cuppen,Marieke Simonis (2015). Lack of Major Genome Instability in Tumors of p53 Null Rats.
Ruben Van Boxtel,Raoul Kuiper,Pim Toonen,Sebastiaan Van Heesch,Roel Hermsen,Alain De Bruin,Edwin Cuppen (2011). Homozygous and Heterozygous p53 Knockout Rats Develop Metastasizing Sarcomas with High Frequency.
Michele Harvey,Hannes Vogel,Danna Morris,Allan Bradley,Alan Bernstein,Lawrence Donehower (1995). A mutant p53 transgene accelerates tumour development in heterozygous but not nullizygous p53–deficient mice.
A Puzio-Kuter,S Laddha,M Castillo-Martin,Y Sun,C Cordon-Cardo,C Chan,A Levine (2015). Involvement of tumor suppressors PTEN and p53 in the formation of multiple subtypes of liposarcoma.
Hannah Beird,Chia-Chin Wu,Davis Ingram,Wei-Lien Wang,Asrar Alimohamed,Curtis Gumbs,Latasha Little,Xingzhi Song,Barry Feig,Christina Roland,Jianhua Zhang,Robert Benjamin,Patrick Hwu,Alexander Lazar,P Futreal,Neeta Somaiah (2018). Genomic profiling of dedifferentiated liposarcoma compared to matched well-differentiated liposarcoma reveals higher genomic complexity and a common origin.
Siddaraju Boregowda,Veena Krishnappa,Jacqueline Strivelli,Christopher Haga,Cori Booker,Donald Phinney (2018). Basal p53 expression is indispensable for mesenchymal stem cell integrity.
Julien Sage (2012). The retinoblastoma tumor suppressor and stem cell biology.
Tania Velletri,Yin Huang,Yu Wang,Qing Li,Mingyuan Hu,Ningxia Xie,Qian Yang,Xiaodong Chen,Qing Chen,Peishun Shou,Yurun Gan,Eleonora Candi,Margherita Annicchiarico-Petruzzelli,Massimiliano Agostini,Huilin Yang,Gerry Melino,Yufang Shi,Ying Wang (2020). Loss of p53 in mesenchymal stem cells promotes alteration of bone remodeling through negative regulation of osteoprotegerin.
Oriol Calvete,Pablo Garcia-Pavia,Fernando Domínguez,Gaelle Bougeard,Kristin Kunze,Andreas Braeuninger,Alex Teule,Adriana Lasa,Teresa Ramón Y Cajal,Gemma Llort,Victoria Fernández,Conxi Lázaro,Miguel Urioste,Javier Benitez (2017). The wide spectrum of POT1 gene variants correlates with multiple cancer types.
Keiji Hirata,Shuichi Kanemitsu,Yoshifumi Nakayama,Naoki Nagata,Hideaki Itoh,Hideo Ohnishi,Hideki Ishikawa,Yoichi Furukawa (2006). A Novel Germline Mutation of MSH2 in a Hereditary Nonpolyposis Colorectal Cancer Patient with Liposarcoma.
Masato Yozu,Pennie Symmans,Michael Dray,Jennifer Griffin,Catherine Han,Daniel Ng,Susan Parry,K Wong (2013). Muir–Torre syndrome-associated pleomorphic liposarcoma arising in a previous radiation field.
Kelly Butnor,Elizabeth Pavlisko,Thomas Sporn,Victor Roggli (2018). Malignant Mesothelioma in Individuals With Nonmesothelial Neoplasms.
M Ferreira (2018). Presence of TERT Promoter Mutations is a Secondary Event and Associates with Elongated Telomere Length in Myxoid Liposarcomas.
Jen-Chieh Lee,Yung-Ming Jeng,Jau-Yu Liau,Jia-Huei Tsai,Hung-Han Hsu,Ching-Yao Yang (2015). Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas.
Lorenza Venturini,Rosita Motta,Alessandro Gronchi,Mariagrazia Daidone,Nadia Zaffaroni (2010). Prognostic relevance of ALT-associated markers in liposarcoma: a comparative analysis.
Rita Lawlor,Nicola Veronese,Antonio Pea,Alessia Nottegar,Lee Smith,Camilla Pilati,Jacopo Demurtas,Matteo Fassan,Liang Cheng,Claudio Luchini (2019). Alternative lengthening of telomeres (ALT) influences survival in soft tissue sarcomas: a systematic review with meta-analysis.
Xiaolei Ren,Chao Tu,Zhenchu Tang,Ruofei Ma,Zhihong Li (2018). Alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas (Review).
Aurora Costa,Maria Daidone,Laura Daprai,Raffaella Villa,Sabrina Cantù,Silvana Pilotti,Luigi Mariani,Alessandro Gronchi,Jeremy Henson,Roger Reddel,Nadia Zaffaroni (2006). Telomere Maintenance Mechanisms in Liposarcomas: Association with Histologic Subtypes and Disease Progression.
Jorge Toro,Lois Travis,Hongyu Wu,Kangmin Zhu,Christopher Fletcher,Susan Devesa (2006). Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978–2001: An analysis of 26,758 cases.
Kristin Baird,Sean Davis,Cristina Antonescu,Ursula Harper,Robert Walker,Yidong Chen,Arthur Glatfelter,Paul Duray,Paul Meltzer (2005). Gene Expression Profiling of Human Sarcomas: Insights into Sarcoma Biology.
Paola Dal Cin,Raf Sciot,Ivo De Wever,Boudewijn Van Damme,Herman Van Den Berghe (1994). New discriminative chromosomal marker in adipose tissue tumors.
David Gisselsson,Michele Hibbard,Paola Dal Cin,Raf Sciot,Bae-Li Hsi,Harry Kozakewich,Jonathan Fletcher (2001). PLAG1 Alterations in Lipoblastoma.
Krzysztof Mrózek,Constantine Karakousis,Clara Bloomfield (1993). High resolution cytogenetic study of adipose tissue tumors: Localization of chromosome 12 breakpoints in lipoma to q15 and in myxoid liposarcoma to q13.3.
Deepika Kanojia,Pushkar Dakle,Anand Mayakonda,Rajeev Parameswaran,Mark Puhaindran,Victor Min,Vikas Madan,Phillip Koeffler (2019). Identification of somatic alterations in lipoma using whole exome sequencing.
Ali Amin-Mansour,Suzanne George,Stefano Sioletic,Scott Carter,Mara Rosenberg,Amaro Taylor-Weiner,Chip Stewart,Aaron Chevalier,Sara Seepo,Adam Tracy,Gad Getz,Jason Hornick,Marisa Nucci,Bradley Quade,George Demetri,Chandrajit Raut,Levi Garraway,Eliezer Van Allen,Andrew Wagner (2019). Genomic Evolutionary Patterns of Leiomyosarcoma and Liposarcoma.
Juan Rosai,Mans Akerman,Paola Dal Cin,Ivo Dewever,Christopher Fletcher,Nils Mandahl,Fredrik Mertens,Felix Mitelman,Anders Rydholm,Raf Sciot,Giovanni Tallini,Herman Van Den Berghe,Wim Van De Ven,Roberta Vanni,Helena Willen (1996). Combined Morphologic and Karyotypic Study of 59 Atypical Lipomatous Tumors.
J Coindre,F Pedeutour,A Aurias,R Tyler (2020). Welldifferentiated and dedifferentiated liposarcomas.
P Hallenborg,S Feddersen,S Francoz,I Murano,U Sundekilde,R Petersen,V Akimov,M Olson,G Lozano,S Cinti,B Gjertsen,L Madsen,J-C Marine,B Blagoev,K Kristiansen (2012). Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation.
Zhao‐jun Liu,Ying Zhuge,Omaida Velazquez (2009). Trafficking and differentiation of mesenchymal stem cells.
Melvin Ambele,Carla Dessels,Chrisna Durandt,Michael Pepper (2016). Genome-wide analysis of gene expression during adipogenesis in human adipose-derived stromal cells reveals novel patterns of gene expression during adipocyte differentiation.
Robin Meyers,Jordan Bryan,James Mcfarland,Barbara Weir,Ann Sizemore,Han Xu,Neekesh Dharia,Phillip Montgomery,Glenn Cowley,Sasha Pantel,Amy Goodale,Yenarae Lee,Levi Ali,Guozhi Jiang,Rakela Lubonja,William Harrington,Matthew Strickland,Ting Wu,Derek Hawes,Victor Zhivich,Meghan Wyatt,Zohra Kalani,Jaime Chang,Michael Okamoto,Kimberly Stegmaier,Todd Golub,Jesse Boehm,Francisca Vazquez,David Root,William Hahn,Aviad Tsherniak (2017). Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells.
Joshua Dempster,Jordan Rossen,Mariya Kazachkova,Joshua Pan,Guillaume Kugener,David Root,Aviad Tsherniak (2019). Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines.
D Ron,J Habener (1992). CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription..
Katarina Engström,Helena Willén,Christina Kåbjörn-Gustafsson,Carola Andersson,Marita Olsson,Melker Göransson,Sofia Järnum,Anita Olofsson,Elisabeth Warnhammar,Pierre Åman (2006). The Myxoid/Round Cell Liposarcoma Fusion Oncogene FUS-DDIT3 and the Normal DDIT3 Induce a Liposarcoma Phenotype in Transfected Human Fibrosarcoma Cells.
Christina Kåbjörn Gustafsson,Katarina Engström,Pierre Åman (2014). DDIT3 Expression in Liposarcoma Development.
N Batchvarova,X Wang,D Ron (1995). Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153)..
Y Lim,B Low,J Lim,E Wong,G Guy (1999). Association of atypical protein kinase C isotypes with the docker protein FRS2 in fibroblast growth factor signaling.
T Kahkonen (2018). Role of fibroblast growth factor receptors (FGFR) and FGFR like-1 (FGFRL1) in mesenchymal stromal cell differentiation to osteoblasts and adipocytes.
Nayan Patel,Sudhesh Kumar,Margaret Eggo (2005). Essential Role of Fibroblast Growth Factor Signaling in Preadipoctye Differentiation.
Le Blanc,S Simann,M Jakob,F Schutze,N Schilling,T (2015). Fibroblast growth factors 1 and 2 inhibit adipogenesis of human bone marrow stromal cells in 3D collagen gels.
P Rogalla,Y Hennig,B Kazmierczak,G Frey,U Deichert,J Bullerdiek (1996). Expression of the HMGI-C gene in uterine leiomyomas and myometrial tissues.
Gilka Gattas,Bradley Quade,Romana Nowak,Cynthia Morton (1999). HMGIC expression in human adult and fetal tissues and in uterine leiomyomata.
Yang Xi,Wanjing Shen,Lili Ma,Ming Zhao,Jiachen Zheng,Shizhong Bu,Shinjiro Hino,Mitsuyoshi Nakao (2016). HMGA2 promotes adipogenesis by activating C/EBPβ-mediated expression of PPARγ.
Helge Thies,Ingo Nolte,Heiner Wenk,Fredrik Mertens,Jörn Bullerdiek,Dominique Markowski (2014). Permanent activation of HMGA2 in lipomas mimics its temporal physiological activation linked to the gain of adipose tissue.
S Battista (1999). The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis.
Azra Ligon,Steven Moore,Melissa Parisi,Matthew Mealiffe,David Harris,Heather Ferguson,Bradley Quade,Cynthia Morton (2005). Constitutional Rearrangement of the Architectural Factor HMGA2: A Novel Human Phenotype Including Overgrowth and Lipomas.
Hyeyun Jung,Won Kim,Do Kim,Yee Cho,Seung Kim,Sung Park,Byoung Park,Heon Lim,Kwang-Hee Bae,Sang Lee (2009). Involvement of PTP-RQ in differentiation during adipogenesis of human mesenchymal stem cells.
Jeong Park,Robert Roeder (2006). GAS41 Is Required for Repression of the p53 Tumor Suppressor Pathway during Normal Cellular Proliferation.
Larissa Pikor,William Lockwood,Kelsie Thu,Emily Vucic,Raj Chari,Adi Gazdar,Stephen Lam,Wan Lam (2013). YEATS4 Is a Novel Oncogene Amplified in Non–Small Cell Lung Cancer That Regulates the p53 Pathway.
Aimee Crago,Nicholas Socci,Penelope Decarolis,Rachael O'connor,Barry Taylor,Li-Xuan Qin,Cristina Antonescu,Samuel Singer (2012). Copy Number Losses Define Subgroups of Dedifferentiated Liposarcoma with Poor Prognosis and Genomic Instability.
Odette Mariani,Caroline Brennetot,Jean-Michel Coindre,Nadège Gruel,Carine Ganem,Olivier Delattre,Marc-Henri Stern,Alain Aurias (2007). JUN Oncogene Amplification and Overexpression Block Adipocytic Differentiation in Highly Aggressive Sarcomas.
Colleen Fullenkamp,Sarah Hall,Omar Jaber,Brittany Pakalniskis,Erica Savage,Johanna Savage,Georgina Ofori-Amanfo,Allyn Lambertz,Stephanie Ivins,Christopher Stipp,Benjamin Miller,Mohammed Milhem,Munir Tanas (2016). TAZ and YAP are frequently activated oncoproteins in sarcomas.
E Seo SOX2 regulates YAP1 to maintain stemness and determine cell fate in the osteo-adipo lineage.
Jan Egan,Michael Barrett,Mia Champion,Sumit Middha,Elizabeth Lenkiewicz,Lisa Evers,Princy Francis,Jessica Schmidt,Chang-Xin Shi,Scott Van Wier,Sandra Badar,Gregory Ahmann,K Kortuem,Nicole Boczek,Rafael Fonseca,David Craig,John Carpten,Mitesh Borad,A Stewart (2014). Whole Genome Analyses of a Well-Differentiated Liposarcoma Reveals Novel SYT1 and DDR2 Rearrangements.
W Tap Evaluation of well-differentiated/dedifferentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization.
C Fletcher,J Bridge,P Hogendoorn,F Mertens (2013). WHO Classification of Tumours of Soft Tissue and Bone.
H Cheng Validation of immature adipogenic status and identification of prognostic biomarkers in myxoid liposarcoma using tissue microarrays.
T Smith,K Easley,J Goldblum (1996). Myxoid/round cell liposarcoma of the extremities. A clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma.
Karen Fritchie,John Goldblum,Raymond Tubbs,Yang Sun,Paula Carver,Steven Billings,Brian Rubin (2012). The Expanded Histologic Spectrum of Myxoid Liposarcoma With an Emphasis on Newly Described Patterns.
P Aman (1992). Rearrangement of the transcription factor gene CHOP in myxoid liposarcomas with t(12;16)(q13;p11).
M Powers Detection of myxoid liposarcoma-associated FUS-DDIT3 rearrangement variants including a newly identified breakpoint using an optimized RT-PCR assay.
A Crozat,P Aman,N Mandahl,D Ron Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma.
N Riggi Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma.
Pedro Pérez-Mancera,Camino Bermejo-Rodríguez,Manuel Sánchez-Martín,Fernando Abollo-Jiménez,Belén Pintado,Isidro Sánchez-García (2008). FUS-DDIT3 Prevents the Development of Adipocytic Precursors in Liposarcoma by Repressing PPARγ and C/EBPα and Activating eIF4E.
M Trautmann FUS-DDIT3 Fusion Protein-Driven IGF-IR Signaling is a Therapeutic Target in Myxoid Liposarcoma.
N Borjigin TLS-CHOP represses miR-486 expression, upregulation of a metastasis regulator PAI-1 in human myxoid liposarcoma.
Silvana Pilotti,Cinzia Lavarino,Alessandra Mezzelani,Gabriella Torre,Fabiola Minoletti,Gabriella Sozzi,Alberto Azzarelli,Franco Rilke,Marco Pierotti (1998). Limited Role of TP53 and TP53-Related Genes in Myxoid Liposarcoma.
B Bode-Lesniewska Relevance of translocation type in myxoid liposarcoma and identification of a novel EWSR1-DDIT3 fusion.
H Zinszner,R Albalat,D Ron (1994). A novel effector domain from the RNA-binding protein TLS or EWS is required for oncogenic transformation by CHOP..
H Kovar,Dr,Mr Jekyll,Hyde (2011). The Two Faces of the FUS/EWS/TAF15 Protein Family.
S Pahlich,L Quero,B Roschitzki,R Leemann-Zakaryan,H Gehring Analysis of Ewing sarcoma (EWS)-binding proteins: interaction with hnRNP M, U, and RNA-helicases p68/72 within protein-RNA complexes.
Kentaro Takahama,Katsuhito Kino,Shigeki Arai,Riki Kurokawa,Takanori Oyoshi (2011). Identification of Ewing’s sarcoma protein as a G‐quadruplex DNA‐ and RNA‐binding protein.
D Dormann Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS.
Brenda Waters,Ioannis Panagopoulos,Elizabeth Allen (2000). Genetic Characterization of Angiomatoid Fibrous Histiocytoma Identifies Fusion of the FUS and ATF-1 Genes Induced by a Chromosomal Translocation Involving Bands 12q13 and 16p11.
Emad Raddaoui,Ludvik Donner,Ioannis Panagopoulos (2002). Fusion of the FUS and ATF1 Genes in a Large, Deep-Seated Angiomatoid Fibrous Histiocytoma.
Hitoshi Ichikawa,Kimiko Shimizu,Rieko Katsu,Misao Ohki (1994). Dual Transforming Activities of the FUS (TLS)-ERG Leukemia Fusion Protein Conferred by Two N-Terminal Domains of FUS (TLS).
Ioannis Panagopoulos,Pierre Åman,Thoas Fioretos,Mattias Höglund,Bertil Johansson,Nils Mandahl,Sverre Heim,Mikael Behrendtz,Felix Mitelman (1994). Fusion of the <i>FUS</i> gene with <i>ERG</i> in acute myeloid leukemia with t(16;21)(p11;q22).
C Storlazzi Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma.
S Huang Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors.
C Turc-Carel Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12).
M Ladanyi,W Gerald (1994). Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor.
T Kwiatkowski,D Bosco,A Leclerc,E Tamrazian,C Vanderburg,C Russ,A Davis,J Gilchrist,E Kasarskis,T Munsat,P Valdmanis,G Rouleau,B Hosler,P Cortelli,P De Jong,Y Yoshinaga,J Haines,M Pericak-Vance,J Yan,N Ticozzi,T Siddique,D Mckenna-Yasek,P Sapp,H Horvitz,J Landers,R Brown (2009). Mutations in the <i>FUS/TLS</i> Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis.
Caroline Vance,Boris Rogelj,Tibor Hortobágyi,Kurt De Vos,Agnes Nishimura,Jemeen Sreedharan,Xun Hu,Bradley Smith,Deborah Ruddy,Paul Wright,Jeban Ganesalingam,Kelly Williams,Vineeta Tripathi,Safa Al-Saraj,Ammar Al-Chalabi,P Leigh,Ian Blair,Garth Nicholson,Jackie De Belleroche,Jean-Marc Gallo,Christopher Miller,Christopher Shaw (2009). Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6.
R Rademakers Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis.
L Yang Subcellular localization and RNAs determine FUS architecture in different cellular compartments.
J Hemminger,O Iwenofu NY-ESO-1 is a sensitive and specific immunohistochemical marker for myxoid and round cell liposarcomas among related mesenchymal myxoid neoplasms.
E Demicco Involvement of the PI3K/Akt pathway in myxoid/round cell liposarcoma.
Y Oda Frequent alteration of p16(INK4a)/p14(ARF) and p53 pathways in the round cell component of myxoid/round cell liposarcoma: p53 gene alterations and reduced p14(ARF) expression both correlate with poor prognosis.
R Davidovic p14(ARF) methylation is a common event in the pathogenesis and progression of myxoid and pleomorphic liposarcoma.
S Sievers Hypermethylation of the APC promoter but lack of APC mutations in myxoid/round-cell liposarcoma.
Katharine Downes,John Goldblum,Elizabeth Montgomery,Cyril Fisher (2001). Pleomorphic Liposarcoma: A Clinicopathologic Analysis Of 19 Cases.
A Oliveira,A Nascimento (2001). Pleomorphic liposarcoma.
J Nishio (2011). Contributions of Cytogenetics and Molecular Cytogenetics to the Diagnosis of Adipocytic Tumors.
H Schmidt (2005). Gains of 13q are correlated with a poor prognosis in liposarcoma.
Ralf Rieker,Stefan Joos,Christine Bartsch,Frank Willeke,Matthias Schwarzbach,Marta Otaño‐joos,Sybille Ohl,Josef Högel,Thomas Lehnert,Peter Lichter,Herwart Otto,Gunhild Mechtersheimer (2002). Distinct chromosomal imbalances in pleomorphic and in high‐grade dedifferentiated liposarcomas.
A Idbaih Myxoid malignant fibrous histiocytoma and pleomorphic liposarcoma share very similar genomic imbalances.
Fredrik Mertens,Christopher Fletcher,Paola Dal Cin,Ivo De Wever,Nils Mandahl,Felix Mitelman,Juan Rosai,Anders Rydholm,Raf Sciot,Giovanni Tallini,Herman Van Den Berghe,Roberta Vanni,Helena Willén (1998). Cytogenetic analysis of 46 pleomorphic soft tissue sarcomas and correlation with morphologic and clinical features: A report of the CHAMP study group.
Kavita Mardi,Neelam Gupta (2011). Primary pleomorphic liposarcoma of breast: A rare case report.
A Klein Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells.
E Macosko Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets.
J Leighton,M Hu,E Sei,F Meric-Bernstam,N Navin (2011). Reconstructing mutational lineages in breast cancer by multi-patient-targeted single cell DNA sequencing.
A Sarvari Plasticity of Epididymal Adipose Tissue in Response to Diet-Induced Obesity at Single-Nucleus Resolution.
W Sun snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis.
M Emont A single-cell atlas of human and mouse white adipose tissue.
C Coombs,T Dickherber,B Crompton Chasing ctDNA in Patients With Sarcoma.
D Braig Genotyping of circulating cell-free DNA enables noninvasive tumor detection in myxoid liposarcomas.
Joanna Przybyl,Lien Spans,Kristen Ganjoo,Nam Bui,David Mohler,Jeffrey Norton,George Poultsides,Maria Debiec-Rychter,Matt Van De Rijn (2022). Detection of MDM2 amplification by shallow whole genome sequencing of cell-free DNA of patients with dedifferentiated liposarcoma.
D Akolkar (2022). Evaluation of patient-specific cell free DNA assays for monitoring of minimal residual disease in solid tumors.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Hannah Beird. 2026. \u201cThe Genomics of Liposarcoma a Review and Commentary\u201d. Global Journal of Science Frontier Research - G: Bio-Tech & Genetics GJSFR-G Volume 22 (GJSFR Volume 22 Issue G2).
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.