## I. INTRODUCTION
- The global scientific community is very much accustomed to the branches like 'chemical thermodynamics' and the 'physics of heat engines' for long many years. The said branches principally deal with the thermodynamic functions like pressure (P), volume (V), heat content (H), internal energy (U), entropy (S), Gibbs free energy (G),....etc.. and the related physical variables like 'time' (t) and 'mass' (m). The demerits of the conventional theories of thermodynamics and the principles of heat engines are described below:
1. The thermodynamic functions except the volume function (V), are all 'abstract' in the sense that their topologies or the geometrical shapes have never been cultivated. The occupied space of a matter is its volume and at ease one can assimilate the concept of what a 'volume' is in the real world. As easy is the understanding of the 'volume', the equal but tougher is the understanding of the other thermodynamic functions like pressure, heat content, internal energy, entropy, the Gibbs free energy, time, mass and the others. While the tripartite definitions (the physics of formation, mathematical expression and the topology or the geometrical shape) do exist for 'volume', however, for the others, mostly we know the mathematical expressions only but their 'physics of evolution' and the 'topologies' are not known to us that much, in the true sense.
2. The geometrical proof of the three 'thermodynamic laws' [1,3,8] have never been put up in the literature in the history of science and the link up of the said laws to the 'space-time' of the universe was never been thought off.
3. The Carnot heat engine utilizes consecutively the 'isothermal' and 'adiabatic' expansion and compressions of a working substance of a thermodynamic system fitted with a piston to yield work from the heat obtained from the source. While the back and the forth movements of a piston are required for a heat engine to work, why such movements of the piston are not initiated only through the isothermal expansion and compressions of the working substance by alternately putting the system on the heat source and the sink? No explanation has been provided.
4. The increase in entropy of the first isothermal expansion process exactly equals the decrease in entropy of the second isothermal process in a Carnot cycle then why it is told that a Carnot Engine does continuously go on generating entropy throughout during its operation.
5. The geometrical profiles of the 'isothermal' and the 'adiabatic' processes are very important for the understanding of the chemical thermodynamics and the physics of the heat engines but those were never drawn up.
Thermodynamics is a subject which is very much related to the physics, mathematics and topology of the 'space - time' of the universe and hence understanding of the principles of thermodynamics and the subject as such is not possible ignoring the said 'space-time' constitution of the universe.
In this article a dedicated effort has been made to make the concepts of thermodynamics very much visible to the readers through the Theory of Space Quantization (TSQ) and by topological or geometrical presentations.
## II. QUANTIZED 'SPACE-TIME' REALM OF THE UNIVERSE
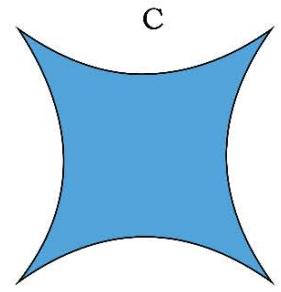
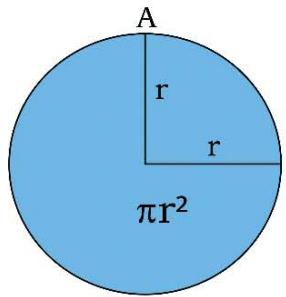
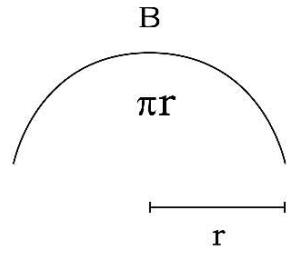
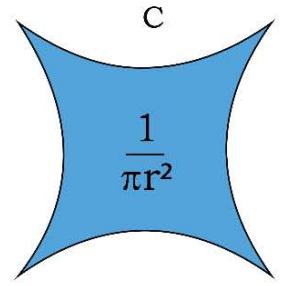
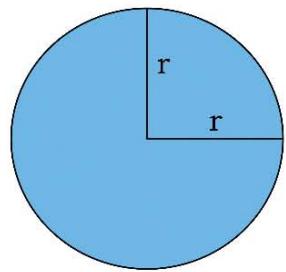
The TTQG/TSQ [1-8] depicted topologies or the geometries of the space quantums are shown pictorially in Figure 1, Figure 2 and Figure 3 below:
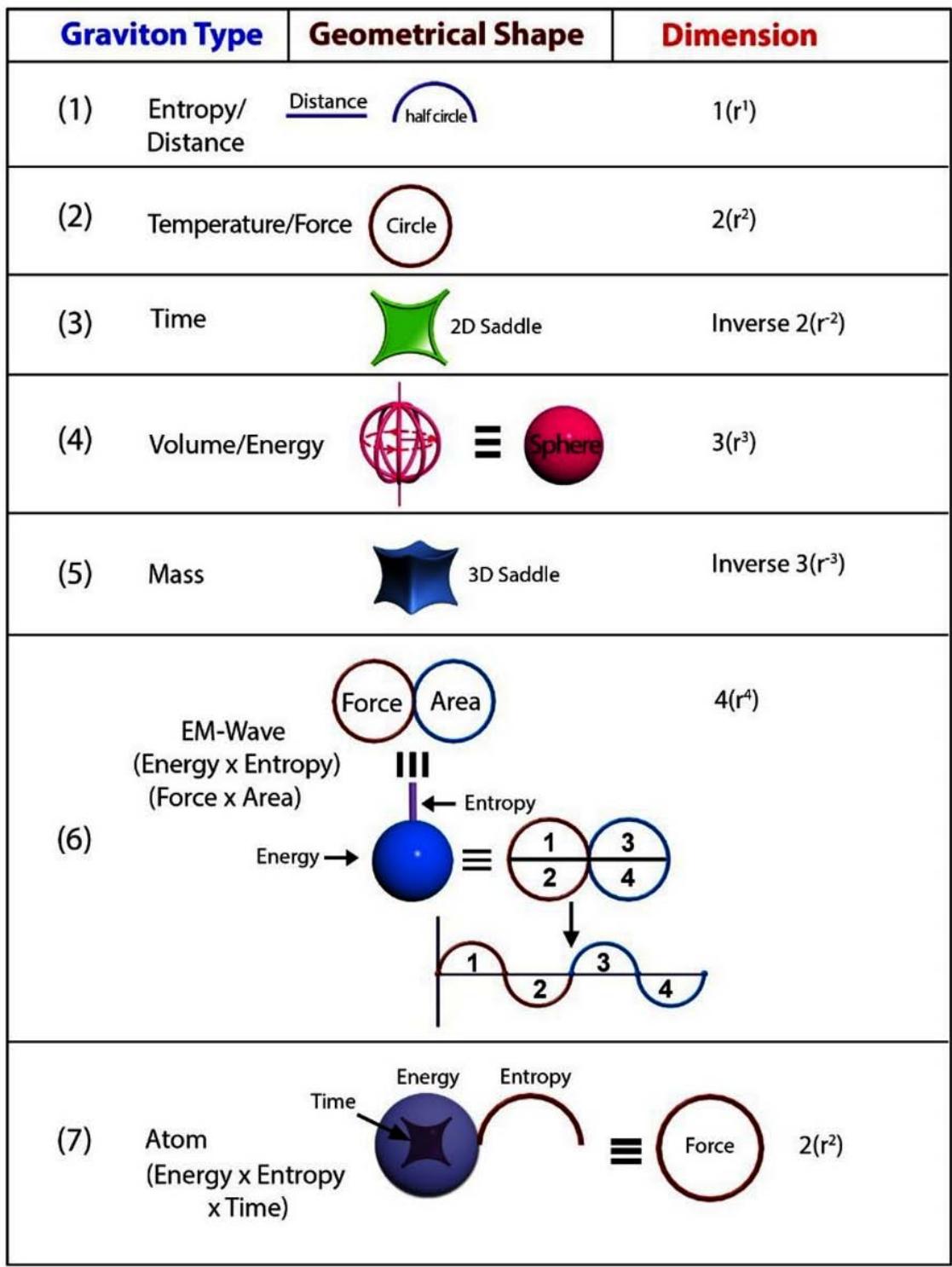 Figure 1: Geometry of Space Quantums of the TSQ
$$
AREA OF CIRCLE = \pi (RADIUS)^{2}
$$
$$
A R E A O F 2 D - S A D D L E = \frac {1}{\pi (R A D I U S) ^ {2}}
$$
 Panel label: I.
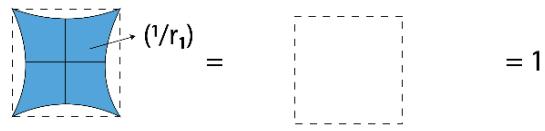
 Panel label: II.
 Panel label: III.
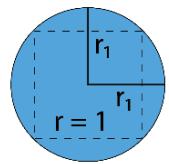
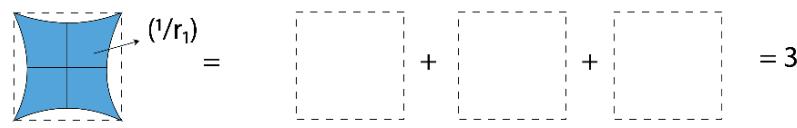 Figure 2: TSQ Presentation of Pressure as a Multiplicative inverse of 'Push Forward Force' and 'Pull Back Force'
$$
\begin{array}{l} P = P R E S S U R E = (P u s h F o r w a r d F o r c e \times P u l l B a c k F o r c e) \\= \text {(A r e a o f C i r c l e} \times \text {A r e a o f 2 D S a d d l e)} \\\end{array}
$$
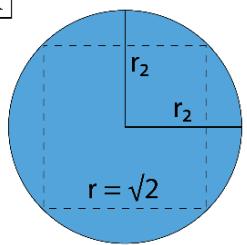
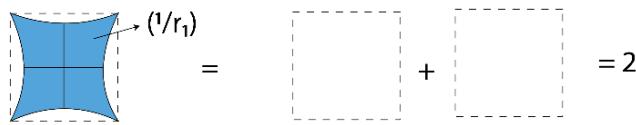
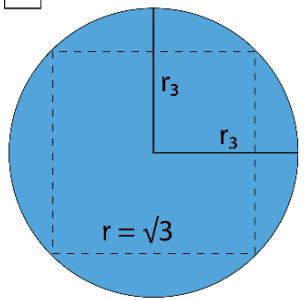
For I $\mathrm{P} = (\pi \times 1 / \pi) = 1 = 1$ No. of Unit Vacuum Space For II $\mathrm{P} = (2\pi \times 1 / \pi) = 2 = 2$ No. of Unit Vacuum Space For III $\mathrm{P} = (3\pi \times 1 / \pi) = 3 = 3$ No. of Unit Vacuum Space
### Integrated Form of Heat
## Differential Form of Heat as Force x Distance
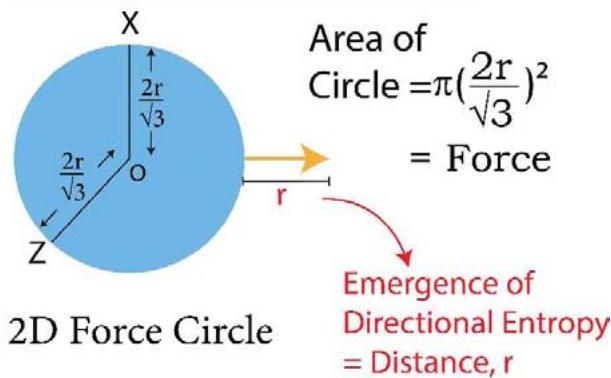 Figure 3: TSQ presentation of 'Integrated' and 'Differential' forms of heat
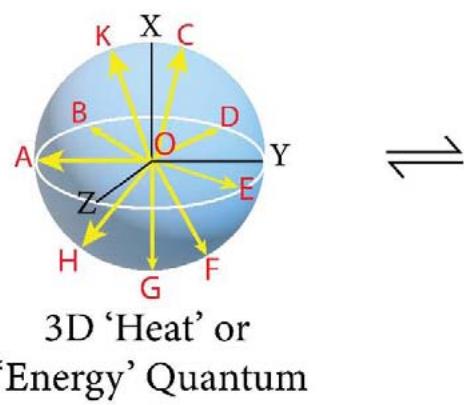
X,Y and Z $\longrightarrow$ 3 Principal Directions in space
OA, OB, OC, OD,... Multi-directional Entropies
$$
O A = O B = O C = r
$$
Volume/Energy of 3D - Sphere $= \frac{4}{3}\pi r^3$
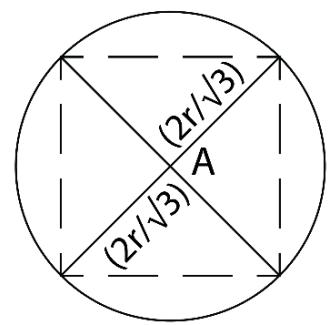
$$
\begin{array}{l} \text{Work} = \text{Force} \times \text{Distance} \\= \pi \left(\frac{2 r}{\sqrt{3}}\right) ^ {2} \times r \\= \frac{4}{3} \pi r ^ {3} \\\end{array}
$$
$$
Energy = Force x Distance
$$
In the TSQ, the following identities of the multivarious space quantums are to be noted:
1. The space quantum are principally two types, i.e., 'direct space quantum' and 'reciprocal or inverse space quantum' or 'push forward space quantum' and 'pull back space quantum'. For example, in TSQ [1], a circle stands for a 'force' physical variable and the inverse of the same (circle) which is a 2D saddle, stands for the 'time' variable of the universe. (Figure 1)
2. 'Entropy' (S) is a line segment and/or a half circle space quantum of direct space. (Figure1). Entropy is basically generating the 'path length' or 'distance'. Any 'distance' in the space is the integral multiple of the length of a 'entropy' quantum.
[The question which could be raised regarding the entropy parameter, how can entropy be a line segment or a curved line segment? Although why it is so, has been explained in the recent publications in triplite manner [1], viz, by the physics of evolution of 'entropy', the underlying mathematics of entropy calculation and the emergence of the topology of entropy. However, there are some very simplistic approach to understand this but the readers of this research article have to come out first or remove from their minds of the abstract definition of entropy as (energy/degree of temperature) presented in the conventional thermodynamics. The index of 'randomness' is 'entropy' but 'entropy' is not the 'randomness'. In TSQ, randomness expresses the magnitude of energy in a '3D spherical shape space quantum'. As the said space quantum increases in size, the energy increases and hence the randomness also increases. To start with a smaller size 'energy quantum' if one goes on increasing its size, the randomness/energy will go on increasing. So the index of randomness is the radius of the '3D sphere space quantum'. If the radius is higher, randomness is higher
and if it be smaller, the randomness is also smaller. So the radius of the sphere (being a line segment) is the index of randomness. This leads one to conclude that 'entropies' are uni-dimensional length space quantums which express the 'degree of randomness'.
Another simple way to learn about entropy parameter may be related to the effect of swimming actions on water. Suppose, two persons, for example, are swimming in a swimming pool side by side. If the speed of swimming (movement of hands) of the said persons be the same, then the person with the higher length of his/her hand will impose more random movements of the molecules of water (and the thrust of the force will reach to the more depth of water) than the person with a shorter length hand. The said first person will displace higher volume of water too.
Another very simple example, one may put forward to make this new concept more explicit, is the correlation of 'height to weight' ratios of the people. Though 'height' is not the 'weight' but the former is the index of the latter, as we all are familiar with. In general, higher the height of a person, higher would be the weight of the person. Higher be the length of the radius of a circle/sphere, higher would be the area/volume and higher would be the entropy and the converse would be true too].
1. 'Temperature' (T) and 'Force' (F) both are represented by a 'circle quantum' of direct space. (Figure1)
2. 'Volume' (V) and 'energy' (E) both are represented by a 'sphere quantum' (Figure 1) or a 'distorted sphere in the form of ellipsoid' [8] of the direct space and under the condition of equilibrium, the energy density of the space is constant such that, $(\mathsf{E} = 3\mathsf{V})$ or $(\mathsf{E} / \mathsf{V} = 3)$ [1-8]. For a thermodynamic process taking place under the condition of constant atmospheric pressure, the energy density would remain constant but the thermodynamic processes taking place under the non-equilibrium conditions (as for example isothermal or adiabatic expansion or compression, where the pressure of the initial states are not same to the atmosphere pressure) since the pressure varies continuously during the said changes, the principle of the constancy of energy density will not be retained).
3. 'Time' is a '2D- saddle' of the reciprocal or the inverse space of the universe. (Figure 1)
4. 'Mass' is a '3D-saddle' of the reciprocal or the inverse space of the universe. (Figure 1)
5. The thermodynamic parameter 'enthalpy' or 'total heat content' (H) being a purely energy parameter is also been represented by a 'sphere or distorted sphere quantum' of the universe.
6. The physical variable 'pressure' in TSQ is a dimensionless parameter as shown in Figure 2. The
geometry of the direct space force quantum swallows the geometry of the reciprocal space force graviton such that the 'pressure' turns out to be multiple only as shown in Figure 2 and the pressure is represented as:
Pressure = (push forward force x pull back force)
7. The 'baryonic matters' (or simply the 'matters' of the universe) or the 'atoms' are the hybrid of three different space quantum, viz, the 'energy' quantum, the 'entropy' quantum and the 'time' quantum and the most logical representation of an atom of a baryonic matter is in the following hybrid form [1]:
Atom of a baryonic matter = (Energy x Entropy x time) As shown in Figure 1 and Figure 4, in a matter, the geometries of energy, entropy and time interact with each other and what is ultimately left is a circle or a quantum of 'force'. As a matter of fact, the atoms vis-à-vis the molecules are the representation of forces of the universe but when they move they acquire 'distance' or 'entropy' and the hybridization takes place between the two in the form of (force x distance) or (force x entropy) and that is the quantum of energy in the form of a 3D sphere since the bi-dimensionality of a force circle being multiplied with a uni-dimensionality of the entropy quantum give rise to a three dimension.
### ATOM OF MATTER
'Energy'
'Entropy'
πr 1
Time'
Dimension $\pi r^2\times 1$
(A/r)
2D Force Circle
(A/r) Swallows C
πr combines with r C
Force Circle Final Form of an 'Atom' Figure 4: TSQ Presentation of an Atom as a Hybrid of 'Energy', 'Entropy' and 'Time'
8. Thermodynamics is a subject which principally deals with 'heat' and 'work'. While we experience the essence of heat in day to day life when we cook, we put on a hair drier to dry up the hair, go to the sea beaches to take sun baths or join warm hands with each other but when it comes to the question of 'work' and that too in the thermodynamic sense (which states that 'work' is not only a simple
algebraic product of force and distance but it is a phenomena of 'order' too), a very clear picture does not simply float in front of our eyes.
The difference between 'disorder' and 'order' is, the former one is multi directional and the latter is unidirectional. While 'energy' or 'randomness' is multi directional, 'work' is uni - directional and in the language of TSQ, the 'energy' or 'heat' is in the integrated or hybrid form and the 'work' is the differential form of 'heat' or 'energy' as shown in Figure 3. In the said figure 3, it is pictorially presented, how a quantum of 'energy' or 'heat' splits into a 'force' quantum and a quantum of 'directional entropy'. While in the heat or energy integrated sphere, the quantum of entropy is multidirectional, the same entropy quantum is unidirectional in the differential form of heat (thermodynamic work).
The function of a heat engine as for example a Carnot engine, is to take up heat from a high temperature source in the form of 3D quantums and to use a portion of the same to initiate the process of the operation of the engine (the first isothermal expansion step) and to release the rest potion of the heat in its differential form of 'force and directional entropy' to the surroundings (in the $3^{\text{rd}}$ isothermal compression step and the $4^{\text{th}}$ adiabatic compression state) such that the 'thermodynamic work in the ordered form' is obtained. The first isothermal step is forcibly initiated but the other three steps, viz, the $2^{\text{nd}}$ adiabatic expansion step, the
3rd isothermal compression step and the final adiabatic compression state take place on their own just by creating the isothermal and adiabatic conditions of the system by altering the position of the system from hot reservoir to cold sink respectively. Although a running car or a running vehicle changes their movements from straight to right or straight to left or vice versa time to time as required but their direction of movement is more or less directional. For this directional movement to happen the heat has to transform to its differential form only and that is what is called the 'thermodynamic work'.
9. While one of the differential form of 'heat' is 'thermodynamic work' as explained in the previous section, the another differential or split-up (or unfolded) form is 'heat' (heat content, H, enthalpy) [1] is in the form of 'internal energy' (U) and 'volume energy' (PV) of the familiar thermodynamic relation, $(\mathsf{H} = \mathsf{U} + \mathsf{PV})$. The pictorial presentation of the said differential form of H is shown in Figure 5 and Figure 6 below:
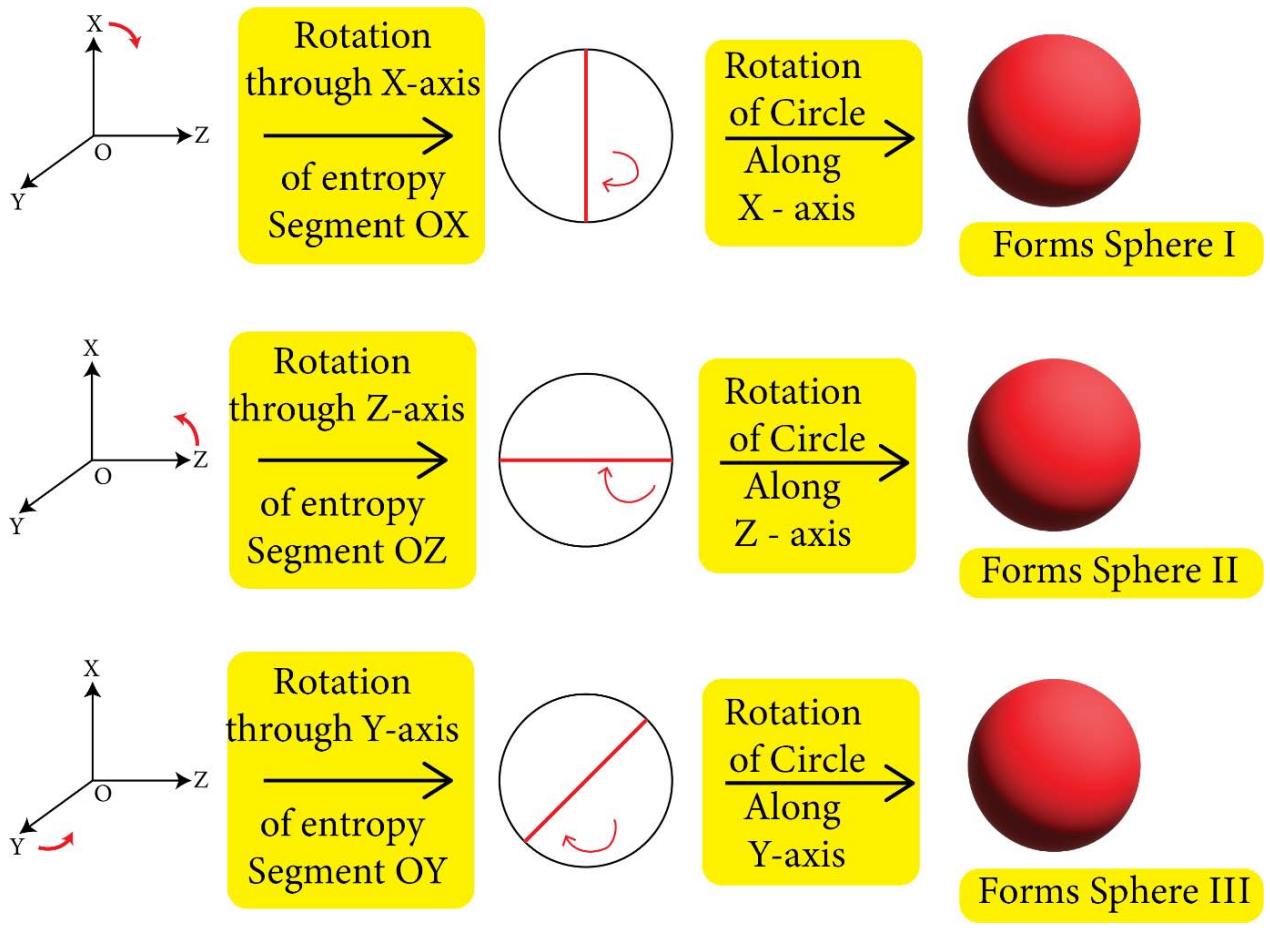 Figure 5: Formation of 3 numbers of 'Volume Quantum' from 3 numbers of 'Entropy Quantum'
2D and 3D Topological Presentation of Heat Content, H
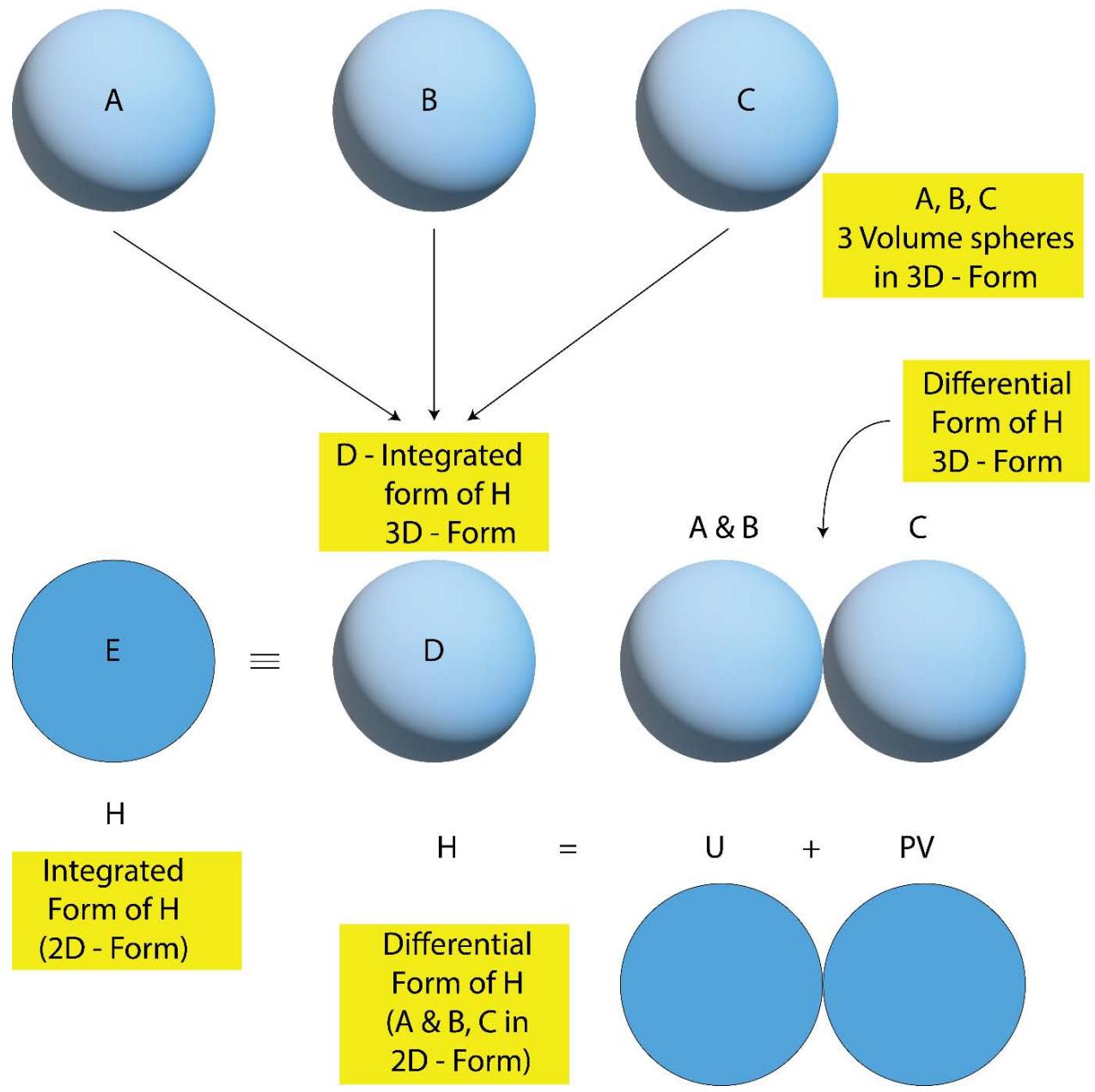 Figure 6: Formation of an 'Energy Quantum' from the Super positions of the 3 numbers of 'Volume Quantum' (as shown in
In Figure 5, it has been shown how an integrated 3D quantum of 'heat or energy' is formed from the rotation of an entropy graviton (line segment) and the rotation of the 'force' circles over the arbitrarily chosen the three principal axis's(X, Y & Z) respectively in the space. So a integrated 3D 'heat or energy' quantum is composed of three numbers of spheres (quantum of volume) basically. Out of the three numbers of quantums of 'volume PV' the two remains in the state of 'superposition' (overlapped with each other) and the third one remains in a perpendicular position to the plane of the other two volume quantums. Now when such a 3D quantum of 'heat' splits into its differential form in two parts, viz, 'U' and 'PV', the two numbers of overlapped volume quantum retain their state of superposition and represents the 'internal energy, U or 2PV' and the perpendicularly oriented one just comes out and is placed in close proximity to the other and represents the 'volume energy, PV'.
$$
Heatcontent = \mathrm{H} = 3\mathrm{PV} = 2\mathrm{PV} + \mathrm{PV} = \mathrm{U} + \mathrm{PV}
$$
$$
(2PV = U, as explained above)
$$
Since P is a multiple only and under the normal atmospheric or equilibrium condition, this multiple P is unity, and the heat content takes the form,
$$
H = 2V + V = 3V
$$
$$
\mathrm {O r} \quad (\mathrm {H} / \mathrm {V}) = 3 \tag {1.1}
$$
In TSQ, $H$ is represented by energy 'E', such that $(E / V) = 3$ or the 'Energy density of the space is constant under the equilibrium condition' [1].
All these are well described through pictorial representation in Figure 6. The volumes of the circles of the U part increase in size when the internal energy increases or decreases in size when the internal energy decreases. On the other hand, the PV part (principally the thermodynamic order creating part) can not only increase or decrease in size homogeneously over all the directions in space but also can be stretched or distorted longitudinally or laterally such that they can encroach the entropy of surroundings and that too directionally and attain the shapes of homogeneous or in homogeneous ellipsoids and would yield the 'thermodynamic work' better. For example if a glass of water (which is kept at ambient temperature under the normal atmospheric pressure) is transferred to a narrower width glass cylinder (at the ambient temperature only and the atmospheric pressure remaining the same), since the average path length of travel of the molecules are increasing and the system is encroaching the space of the surroundings, and are forced to move in ordered and directional manner, the directional entropy would increase and creating such situations one can generate more 'thermodynamic work' and this will be elaborated later in this article.
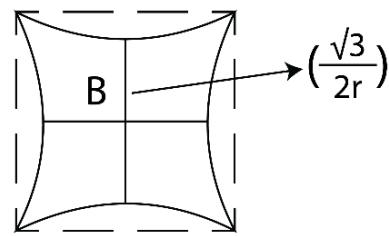
The TSQ defines temperature (T) as a force circle which is dispersive by its nature and is labelled as a 'push forward quantum' of the universe and is expanding the space. On the contrary, the 'time quantum, t' is a 'pull back force quantum' which is represented by a '2D saddle' which is attractive by its nature and is holding back the space of the universe. In the state of equilibrium T and t are multiplicative inverse to each other such that it is being mathematically expressed as:
$$
\mathrm{T}t=1
$$
The physical significance of equation (1.2) is, when a 'quantum of temperature (T)' interacts with a 'quantum of time (t), what is formed is a 'empty space $\pi$ quantum' or a 'empty space $\pi$ graviton' as shown in Figure 7.
$$
^ {\prime} T t = 1 = 1 \text{Numberof} \pi - \text{Graviton} ^ {\prime}
$$
'TEMPERATURE CIRCLE' T
$$
\begin{array}{l} \text{Temperature} \\= \pi \left(\frac{2r}{\sqrt{3}}\right) ^ {2} \\= \frac{4}{3} \pi r ^ {2} \\\end{array}
$$
'TIME 2D - SADDLE' 't'
$$
Time = \frac{1}{\pi \left(\frac{2 r}{\sqrt{3}}\right)^{2}} = \frac{3}{4 \pi r^{2}}
$$
 Panel label: C D.
 Figure 7: Formation of a
$\pi$ -quantum' from the interaction of a 'Temperature Quantum' and a 'Time Quantum'
$\pi$ -SPACE QUANTUM or
$\pi - \text{GRAVITON}$
The beauty of the 'space quantum's are, they are inter-controvertible to each other. In Figure 9 and 10 below, the inter-convertibility of the various quantums among themselves are being shown:
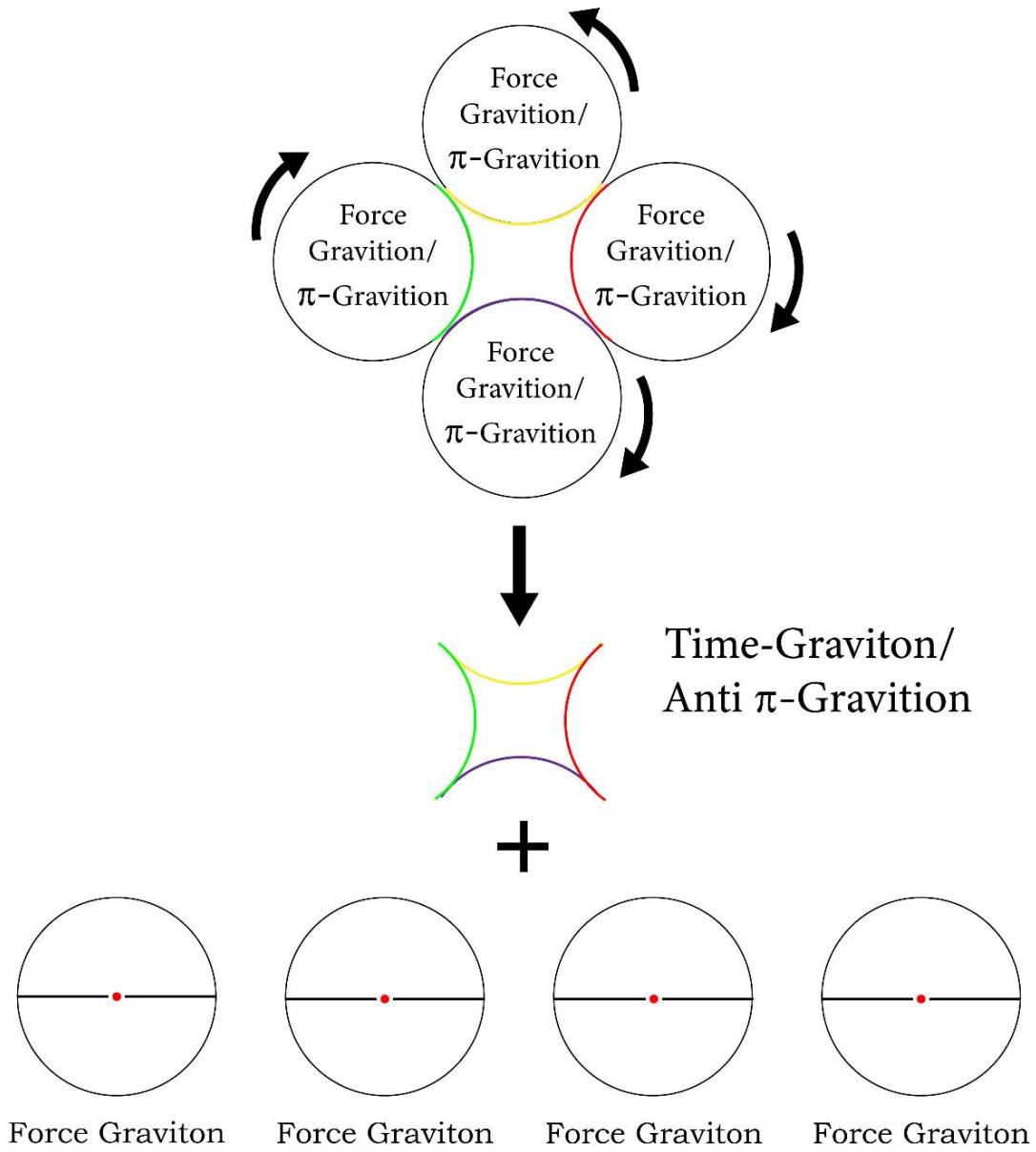 Figure 8: The inter-convertibility of 'empty
$\pi$ quantum' and 'empty anti- $\pi$ quantum' or a 'Force Quantum' and a 'Time Quantum'
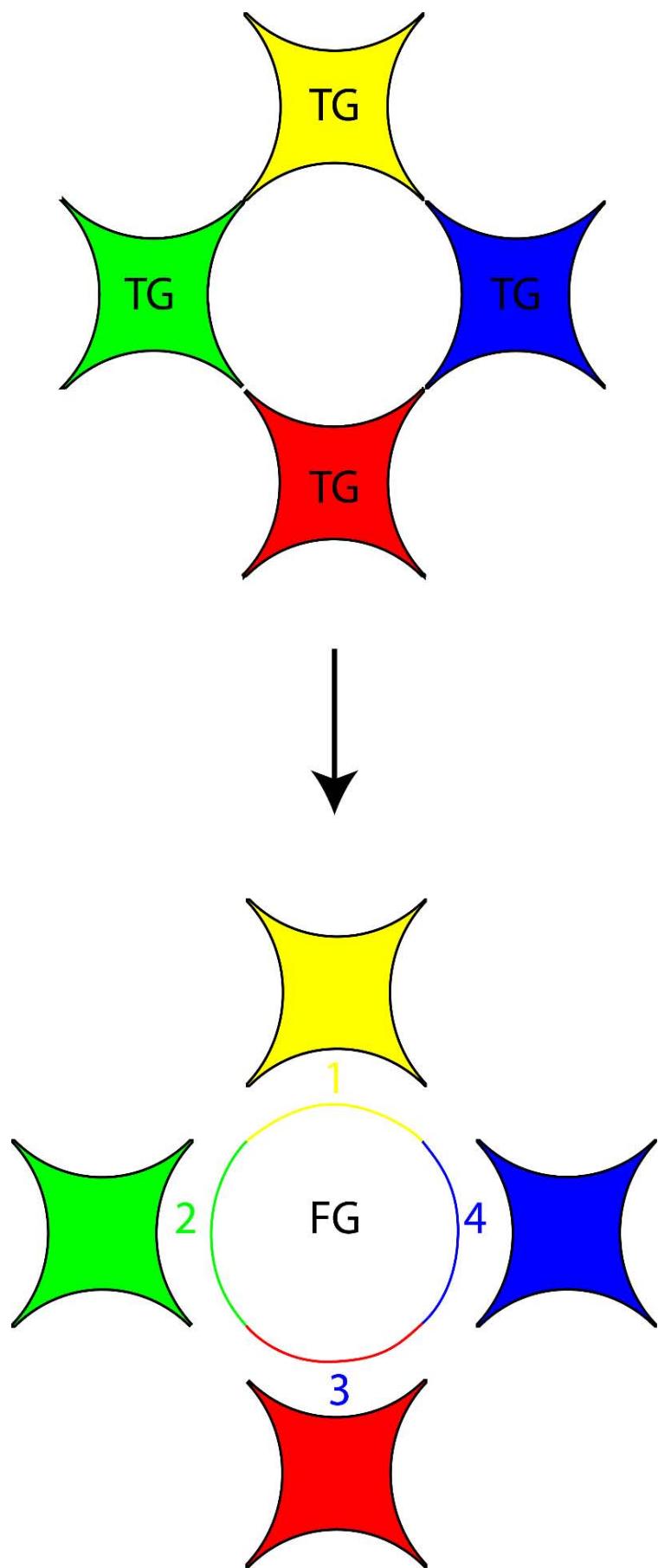 Figure 9: The inter-convertibility of 'empty anti
$\pi$ quantum' and 'empty $\pi$ quantum' or a 'time quantum' and a 'Force Quantum' (FG - Force quantum, TG - Time Quantum) In TSQ it has been shown that when a quantum of 'mass' (m) interacts with a quantum of volume (V), three numbers of 'empty $\pi$ quantum' are generated [1,4]. The 'empty $\pi$ quantum' are the carriers or packets of the push forward space quantums of the universe like 'entropy', 'force', 'temperature', 'volume', 'energy'.etc and the 'empty anti- $\pi$ quantum' are the carriers or packets of the 'pull back quantum's of the universe like 'order', 'time', 'mass' etc. While three numbers of 'empty $\pi$ quantum' are generated when mass interacts with volume, nine numbers of 'empty $\pi$ quantum' are evolved when the same (the mass quantum) interacts with a quantum of 'energy'. So these are mathematically expressed in TSQ as
$$
\mathrm {m V} = 3 \tag {1.3}
$$
$$
mE = 9
$$
a) The Underlying Logic of the Emergence of the Helmholtz's Free Energy (F) and the Gibbs Free Energy (G) Parameters as the Key Indices of the 'Thermodynamic Work'
We are very to very much familiar with the famous following two mathematical equations of thermodynamics
$$
G = H - T S \tag {2.1}
$$
$$
F = U - T S \tag {2.2}
$$
The following two points to be noted in regard to the incompleteness of the above said equations and another one important theorem of thermodynamics:
1. The equations have just been put forward in thermodynamics without their proper derivations.
2. The parameter 'S' in these equations have been referred to as 'entropy parameter' but it has not been clearly spelt out that whether it is the entropy of the 'system' only or only the entropy of the 'surroundings' or the entropy of the 'system plus the surroundings.
3. When it comes to the question of surroundings, it is to be noted very much, that the surroundings beyond a well mapped system is enormous and vast and so although it might be possible to assess the entropy of the system but how can one measure or assess the entropy of the vast surroundings? So keeping this subject at total dark, by some mathematical arguments (those are tedious though), the criteria of spontaneity of any process occurring in nature has been projected as 'For a spontaneous process, $\left( \Delta S_{\text{system}} + \Delta S_{\text{surr}} \right) > 0'$. $\Delta S_{\text{system}}$ and $\Delta S_{\text{surr}}$ stand for the changes in entropy of the system and the surroundings respectively during a change of a thermodynamic state.
In regard to TSQ, the parameter TS is:
$$
T = Temperature = Push Forward Force
$$
$$
S = entropy = Distance
$$
$(TS) = (Force \times Distance) = Total$ energy involved in the process (7)
So if a gas is continued expanding under constant pressure condition, both the 'T' and the 'S' go on increasing and if the said 'S' is considered to be the 'entropy of the system', the 'TS' or the energy involved in the process would go on increasing towards an infinite magnitude and the surroundings would have simply starved of energy or would have simply frozen out. So 'S' cannot be the entropy of the system only.
The conventional concept of 'system' and 'surroundings' needs to be rebuilt for the following valid logic. If the case of an expansion of a gas is considered under the isothermal conditions, the volume of the gas goes on increasing from the initial volume and the system starts encroaching the space of the surroundings. Under such situation the system and surroundings both are gradually becoming the integral parts to each other and they behave as a composite one as ('system + surroundings'). To make this new but very tangible concept more explicit, the case of a spontaneous expansion of a gas can be cited. Suppose a cylinder (fitted with a piston) filled with a certain amount of a heated gas at high pressure is hold at a certain height of the cylinder, by putting a weight on the piston. In fact, this is a non-equilibrium situation and let the value of the entropy of the gas/system be 'X'. Now if the weight which is put on the piston, is being removed at any instant and the gas is allowed to expand under isothermal conditions on its own, the piston will move upwards but would stop after reaching a certain height above its original height (when the pressure of the gas equals the atmospheric pressure). Now if the magnitude of the entropy had increased to a level of 'Y' (from its original value of X), this new increased value of entropy would have to be considered as the entropy of ('system + surroundings') because the system had encroached some space of the surroundings and the system and the surroundings together would be behaving as a 'composite entity' of the universe. Now let us assess the situation as under as shown in Figure 10:
Change in Entropy of System + Surroundings > 0
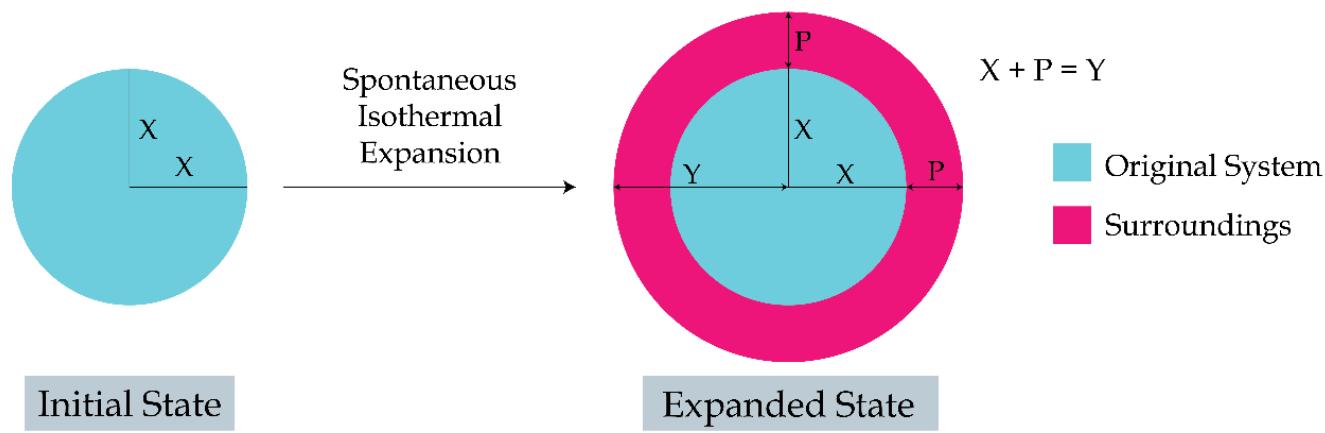 Figure 10: TSQ Concept of the 'surroundings' and the thermodynamic Condition of 'Spontaneity' of a physical Process in the Nature
Entropy of the System $= X$
Entropy of the Surroundings $= 0$
Entropy of System + Surroundings
$$
\begin{array}{l} = X + 0 \\= X \\\end{array}
$$
Entropy of the System $= X$
Entropy of the Surroundings $= \mathrm{P}$
Entropy of System + Surroundings
$$
\begin{array}{l} = X + P \\= X \\\end{array}
$$
Change in Entropy of the System + Surroundings $= \mathrm{Y} - \mathrm{X} > 0$
So,
$\Delta (\mathrm{S}_{\mathrm{system}} + \mathrm{S}_{\mathrm{surroundings}}) > 0$
Entropy of the system at the initial state $= X$
Entropy contribution of the surroundings at the initial state $= 0$
Entropy of the (system + surroundings) at the initial state = X
Entropy of the (system + surroundings) at the final state = Y
Change in Entropy of the (System + Surroundings) between the final and initial state is = (Y - X)
Since $Y > X$, it turns out in mathematical form $\Delta(S_{\text{System + Surroundings}}) > 0$ and which is not being true for only a spontaneous expansion of a gas as cited above but is being true for any spontaneous process occurring in nature and the conventional expression of spontaneity (in the following mathematical form) is not being true,
$$
\left(\Delta_{\text{System}} + \Delta S_{\text{Surroundings}}\right) > 0
$$
Based on the above said logics and the new proposed concepts, the true derivations of the
Helmholtz's Free energy function (F) and the Gibb's free energy function (G) are being illustrated here:
A reversible isothermal expansion of an ideal gas is characterized by,
1. Heat input (from the surroundings) = Work done (by the system),
2. Multi directional entropy is being generated in the system.
So in an isothermal process, the volume energy 'PV' remains constant throughout the entire process since whatever the heat enters the system, is fully utilized as so called work. This means the 'volume energy, PV' remains untouched. So, in an isothermal process, the energy is provided by the 'internal energy or 2PV 'part and the entire 2PV amount of energy can be utilized. However, the surroundings compensate for the same amount of energy (2PV), such that the
temperature/internal energy remains constant through out the entire process.
So, the total energy involved in the process, as explained above in the form 'TS'
$$
T S = 2 P V + 2 P V = [ \text {U t i l i z a t i o n o f i n t e r n a l e n e r g y (2 P V)} ] + [ \text {E n e r g y c o m p e n s a t i o n o f t h e s u r r o u n d i n g s , (2 P V)} ]
$$
At this juncture, a new thermodynamic state function, $F$, called 'Free Energy' was introduced as, $(F = -2PV)$. The logic behind this is the $(2PV = U =$ internal energy of the system) amount of internal energy theoretically can be fully converted to 'work' (in the form of 'force' and 'directional entropy') and the minus sign is put to signify that the said 2PV or $U$ amount of heat or energy would have to leave the system to produce the said 'thermodynamic work'. So $F$ (in the form of -2PV) expresses that how much of maximum work that could be extracted from the system and 'F' emerged as the 'measure of the capability of the work of a system or the maximum work function' and that too under the isothermal condition. For example, if the free energy, $F$, of any two arbitrary chosen systems be, $(-X)$ and $(-Y)$ respectively and $X > Y$, then the first system will be in a position to do more work than the second one.
Now the above equation can be rearranged as,
$$
-2\mathrm{PV} = 2\mathrm{PV} - \mathrm{TS}
$$
$$
Since (2\mathrm{PV}) = \mathrm{U} \text{and} (-2\mathrm{PV}) = \mathrm{F}, \quad \mathrm{F} = \mathrm{U} - \mathrm{TS} \tag{2.4}
$$
So the equation (2.4) is the famous equation of Helmholtz's but to keep in mind that 'S' stands for the 'entropy of system + surroundings' and otherwise which such an equation could never be formed.
Whether it be the free energy parameter $F$ or the free energy parameter $G$, the readers of this article should put well in their mind the following concept of the free energy parameters in regard to mechanical work:
The term 'PV' is there in both F and G and PV = (pressure x Volume) = (Force x volume)/(area) = (Force x distance) = (Force x directional entropy) (in case of thermodynamic work). When a system is resting in equilibrium at one or the other thermodynamic state, the 'free energy parameters' do not carry any significance truly but the moment it starts changing its own thermodynamic state (as for example, isothermal expansion or compressions), the free energy parameter appears at the scenario. For the said changes to take place, some 'force' be it smaller or be it bigger, has to act in the system and under this situation, how much of directional entropy, the system can generate [encroaching the surrounding space in the form of, (entropy of the system + surroundings)] is the measure of its free energy. Higher the magnitude of the product of the 'force' and 'directional entropy' higher is the amount of the thermodynamic work and more negative would be the free energy.
The above said concept should not be kept limited to only the mechanical work under isothermal condition but it should be a generalized concept for any isothermal change, since the work done by a system is the outcome of the, i) energy of movements of the multi various space quantums, i.e., whether the energy associated with the said movements are higher or lower in magnitude, ii) the pattern of the movements, i.e., whether the pattern of movements are 'multi directional' or 'directional' or both and iii) the 'cohesiveness and dispersive character of the system', i.e., the balancing of push forward and pull back quantums. For any type of work output, for example, electrical, mechanical, chemical, diffusional, rheological, all the above said factors play the major roles.
The Gibbs free energy equation is derived below:
For an expansion of a gas taking place under the condition of constant pressure, both the 'volume energy, PV' and the 'internal energy 2PV or U' increases. So, at any instant, the involvement of 'total energy of the system plus surroundings' (TS) is the sum of:
1. 'volume energy, 'PV', surrounding's compensated, (the system exhausts its own volume energy 'PV' and the surroundings compensates for the same and which is distributed partly to the 'internal energy compartment, U' and partly to the 'volume energy compartment, PV')
2. 'internal energy 2PV'
3. 'the volume energy, 'PV'.
So
$$
T S = 2 P V + P V + P V
$$
$$
Or ext{TS} = ext{U} + ext{PV} + ext{PV}
$$
$$
Or\quad\mathrm{TS} = \mathrm{H} + \mathrm{PV} \left(\mathrm{since}\\mathrm{H} = \mathrm{U} + \mathrm{PV}\right)\tag{2.5}
$$
Now the Gibbs free energy parameter has been introduced as $G = (-PV)$ and hence the above equation takes the form:
$$
TS = H - G
$$
$$
\mathrm{Or}\quad G=H-TS\tag{2.6}
$$
The criteria of spontaneity of physical or chemical processes occurring in nature are usually mapped in the conventional chemical thermodynamics by the changes in $\Delta F$ and $\Delta G$ respectively and are represented as following:
For the processes occurring under isothermal conditions and the said processes are mechanical ones,
$$
(\Delta F) _ {T, V} < 0 (T, V \text{remainconstant}) \tag{2.7}
$$
For the processes occurring under isobaric conditions and the said changes are chemical ones,
$$
(\Delta G) _ {P, T} < 0 (P, T \text{remainingconstant}) \tag{2.8}
$$
As a matter of fact, such conclusions are not much logical as has been discussed in recent publications [1] in detail and is not much discussed here. The main points to note regarding the non-appropriateness of the aforesaid criteria of spontaneity are the following:
1. Regarding the spontaneity criteria of $(\Delta F)_{T,V} < 0$, (in case of a mechanical change), the boundary between the system and the surroundings has to move and as a result of that, V cannot remain constant for a homogeneous expansion or compression of a system over all the directions in space. The only situation under which a system retains the constancy of volume but the above said boundary moves is, the system has to expand or contract longitudinally or laterally such that the
Changes in the magnitude of volume (longitudinal/lateral directions) of the expansion/contraction processes counterbalance each other such that the total volume remains constant. These belong to the category of self-ordering phenomena of the molecules, but such types of processes rarely take place spontaneously except for the self-ordering of molecules of very rigid nature to form liquid crystals.
2. Regarding the spontaneity criteria of $(\Delta G)_{P,T} < 0$, (in case of a chemical change or the physical intermixing of two or more substances), since most of them are either exothermic or endothermic, they cannot be considered as true isothermal ones. Also, it has been explained in another publication [4] with proper reasoning that the free energy parameter $G$ is a misfit parameter for chemical reactions and consequently assessing the criteria of spontaneity of chemical reactions through $(\Delta G)$ is also not justified. However, for the cases of physical mixing of two substances (liquid or gas), the $(\Delta G)$ data of the processes can be utilized for determining the spontaneous/ non-spontaneous characters of the said mixing processes.
3. The two equations of $F$ and $G$ can be written in regard to TSQ are:
$$
F = (- 2 P V) = U - T S = (2 P V) - T S \tag {2.9}
$$
$$
G = (- P V) = H - T S = (U + P V) - T S = (2 P V + P V) - \quad T S = (P V) - (T S - 2 P V) = (U / 2) + (F) \tag {2.10}
$$
The magnitudes of F and U are being the same (minus sign in F signifies that -2PV heat is leaving the system). This shows that the internal energy U, under isothermal condition, is fully utilized to produce only mechanical or both mechanical and non- mechanical work and it leaves the system as 'F'. What is then the difference between F and U then? The difference is, the latter one is the integrated form of heat (in the form of force and multi directional entropy) and the former one is the differential form heat, same in magnitude to that of U, which leaves the system(in the form of force and directional entropy) and which is called the 'thermodynamic work'.
In case of $G$, as shown above in equation (2.10), only a half part of the internal energy is utilized, and which leaves the system to perform non-mechanical work one and that too in the form of 'force' and 'directional entropy'.
Equation (2.10) can be broken up in the following manner to derive a very interesting and significant correlation equation between F and G:
$$
\begin{array}{l} G = (U / 2) - (T S - 2 P V) \\= (U / 2) - T (S - 2 P V / T) \left[ (P V / T) = (\text {E n e r g y} / \text {T e m p e r a t u r e} \quad (\text {F o r c e}) = \text {D i s t a n c e} \right. \tag {2.10a} \\\end{array}
$$
As per TSQ,
$$
\text{when} \quad P = 1 (\text{constantpressure}), (2 \mathrm{P V} / \mathrm{T}) = (2 \mathrm{V} / \mathrm{T}) = 2 [ (4 \pi \mathrm{r} ^ {3} / 3) / (4 \pi \mathrm{r} ^ {2} / 3) ] = (2 \mathrm{r}) \tag{2.10b}
$$
Equation (2.10a) can be rewritten as, [considering(S-2r) as reduced entropy, $S_R$ and (U/2) as reduced internal energy $U_R$ (= U/2)
$$
G = (U/2) - T (S - 2 r) = (U/2) - T (S_R) = (U/2) - T S_R
$$
$$
\mathrm {O r}, \quad \mathrm {G} = \mathrm {U} _ {\mathrm {R}} - \mathrm {T S} _ {\mathrm {R}} \tag {2.10c}
$$
And
$$
F = U - T S (k n o w n t o u s)
$$
So, both F and G could be expressed in terms of internal energy/reduced internal energy, temperature and entropy/reduced entropy. Now if figure 6b is looked into one would find that in the differential form of 'H', the ratio of U to PV is $= (2\mathrm{PV} / \mathrm{PV}) = 2$. Whenever a thermodynamic process would occur under constant pressure conditions, the said ratio of U to V have maintained at the level of 2:1. Now as per equation (2.10),
$$
G = F + (U/2) = (-2PV) + (U/2) = = (-2PV) + (-2PV/2) = = (-2PV) + (-PV)
$$
The first part of equation (2.10d) corresponds to 'internal energy' part and the second part corresponds to the P-V part. The ratio of U to PV is $(= -2\mathrm{PV} / -\mathrm{PV}) = 2$
While, in an isothermal expansion process, the heat enters the system and is being partly converted to work, (except for ideal gas where full conversion of heat to work occurs), in the constant pressure expansion process, a part of the heat which enters the system is converted to work and the rest part is being evenly distributed between the 'U' part and the 'PV' part such that the ratio of U to PV remains to be in the ratio of 2:1, since otherwise which, the pressure, P, would not remain constant. For carrying out any thermodynamic process under constant pressure condition the thermodynamic condition one can put is the constancy of the U to PV ratio throughout the entire process.
For the isothermal and adiabatic expansion processes at the initial states (the initial states do not remain in equilibrium with the surroundings since $P$ remains higher), the energy densities ( $U/V$ and $H/V$ ) remain higher (more than 2 and 3 respectively or $U$ and $H$ ). As the expansion process progresses, the said ratios fall monotonically and ultimately levels off towards (2 & 3 respectively) the completion of the processes. In contrast to this, the ( $U/V$ ) and ( $H/V$ ) remains to be 2 and 3 for the expansions occurring under constant pressure condition.
The definition of a spontaneous process or what a spontaneous process is, has never been clearly spelt out in thermodynamics. The main characteristic of spontaneous processes (physical, chemical or any other processes) are, when they are initiated a nonequilibrium situation is evolved instantaneously and the said processes are completed when some or the other 'equilibrium state' is reached [1]. In the event of the initiation of a spontaneous chemical reaction, the forward reaction begins sooner or later as the reactants are mixed and it is for sure is a situation of 'nonequilibrium'. Then the backward reaction starts and when the rate of forward reaction equals the rate of backward reaction, the 'equilibrium state' is attained. One can think about a physical process too in context to this. Suppose, a person is holding a rubber ball in his hand at a certain height above the ground level. When the person just releases the ball by losing his fingers, the rubber ball starts falling due to gravity towards the ground level and instantaneously a situation of 'non- equilibrium state' is evolved. When the rubber ball hits the ground, it bounces several times and ultimately it comes to position of rest and the 'equilibrium state' is being reached. So the true definition of a spontaneous process in nature is 'Either they start on their own or they are initiated with the onset of a non - equilibrium state and that ultimately converges to a state of equilibrium on their own'.
The view of TSQ in very plain and simple form is, at the beginning of a spontaneous process, the product of the 'push forward force' (T) and the 'pull back force' (t) remain in a state of 'non-multiplicative' to each other but when it reaches the state of equilibrium the product of T and t turns out to be multiplicative to each other.
For 'non-equilibrium state' either $T_{t} > 1$ or $T_{t}< 1$ and for a equilibrium state $T_{t} = 1$. The TSQ driven 'rule of thumb for the spontaneity of any processes in the nature is, they are auto or externally initiated (with either $T_{t} > 1$ or $T_{t} < 1$ ) and converge to $T_{t} = 1$ on their own'. This criteria of spontaneity is very to very much universal, absolute and sacrosanct in the sense that it is free of the conditional criteria of spontaneity (as for example, constant temperature, constant volume, constant pressure..etc..) as expressed in the forms of $(\Delta G)_{P,T} < 0$ or $(\Delta F)_{T,V} < 0$, in conventional chemical thermodynamics.
To properly understand the 'free energy' and 'available thermodynamic work' of a system, one needs to understand the 'topology' or the 'geometrical profile' of the 'isothermal' and the 'adiabatic process' [1]. We are familiar with the dictum in chemical thermodynamics like 'isothermal expansion processes are entropy generating (of the system) by their nature' and 'adiabatic expansion processes are iso-entropy or non-entropy generating (of the system) by their nature'. However, chemical thermodynamics had remained silent for years about what are happening to the 'entropy of the surroundings' (whether the entropy increases, decreases or remain to be constant) for the above said isothermal and adiabatic expansion processes. Also, as well, what are the types of entropy (multidirectional or directional) those are associated with the isothermal and adiabatic processes have never been spelt out too. TSQ reveals all these indeed and the following points to be noted:
1. In an isothermal expansion process the entropy generation takes place homogeneously in the 'system + surroundings' over all the directions in space (which is multi-directional one).
2. In an adiabatic expansion process a part of the multidirectional entropy of the 'system' is transformed to the directional entropy and some entropy of the surroundings is also captured and as a result of that, the entropy of the 'system + surroundings' increases.
3. In the case of an isothermal expansion process the initial volume of the system in the form of a homogeneous sphere enlarges to a bigger size homogeneous sphere (encroaching the space of the surroundings over all the directions).
4. In the case of an adiabatic expansion process the initial volume of the system in the form of a homogeneous sphere enlarges to a bigger size inhomogeneous sphere or ellipsoid type (encroaching the space of the surroundings on an average over a specific direction).
5. Without understanding the 'geometrical profiles' of the isothermal and adiabatic expansion and compression processes, one cannot truly understand the principles of the work ability of the heat engines.
The geometrical profiles of typical isothermal 'expansion - compression' and adiabatic 'expansion-compression' are shown in Figure 11a, 11band Figure 12 below:
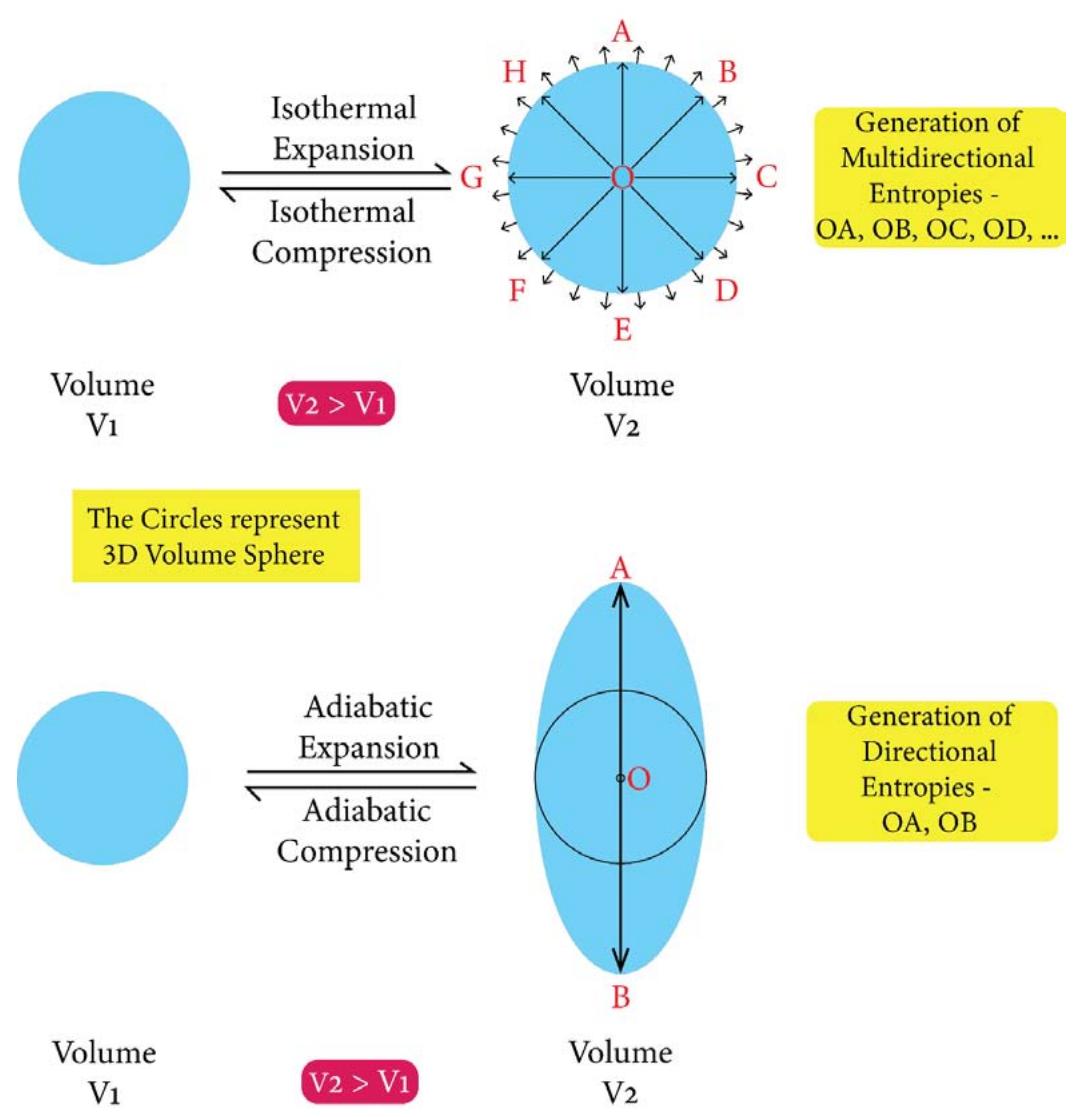
2D Presentation of the Geometric Profiles of the Isothermal and Adiabatic Processes Figure 11a: Topological Presentation of Typical 'Isothermal expansion-compression' and 'Adiabatic expansion-compression' Processes
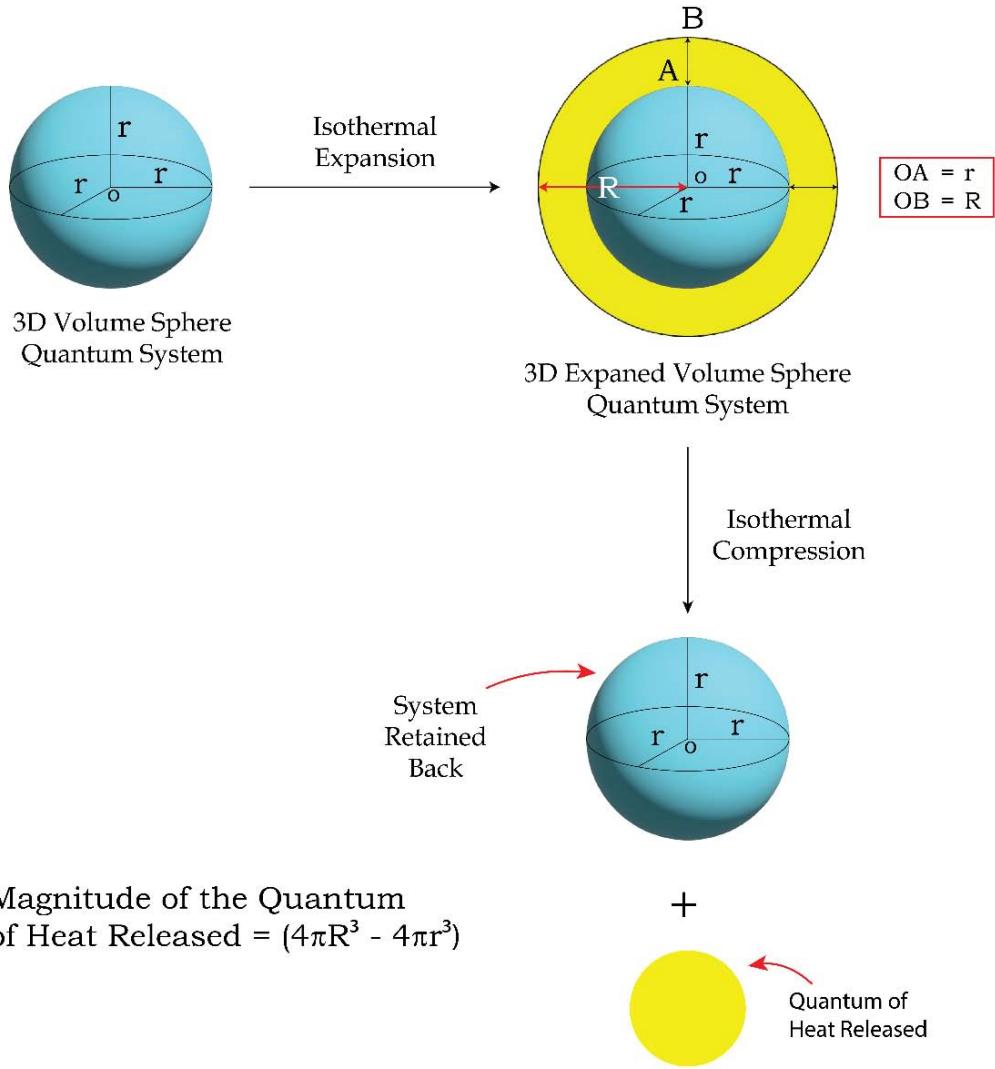
Figure 11b: Quantum Level Presentation of a Typical 'Isothermal Expansion- Compression' Process
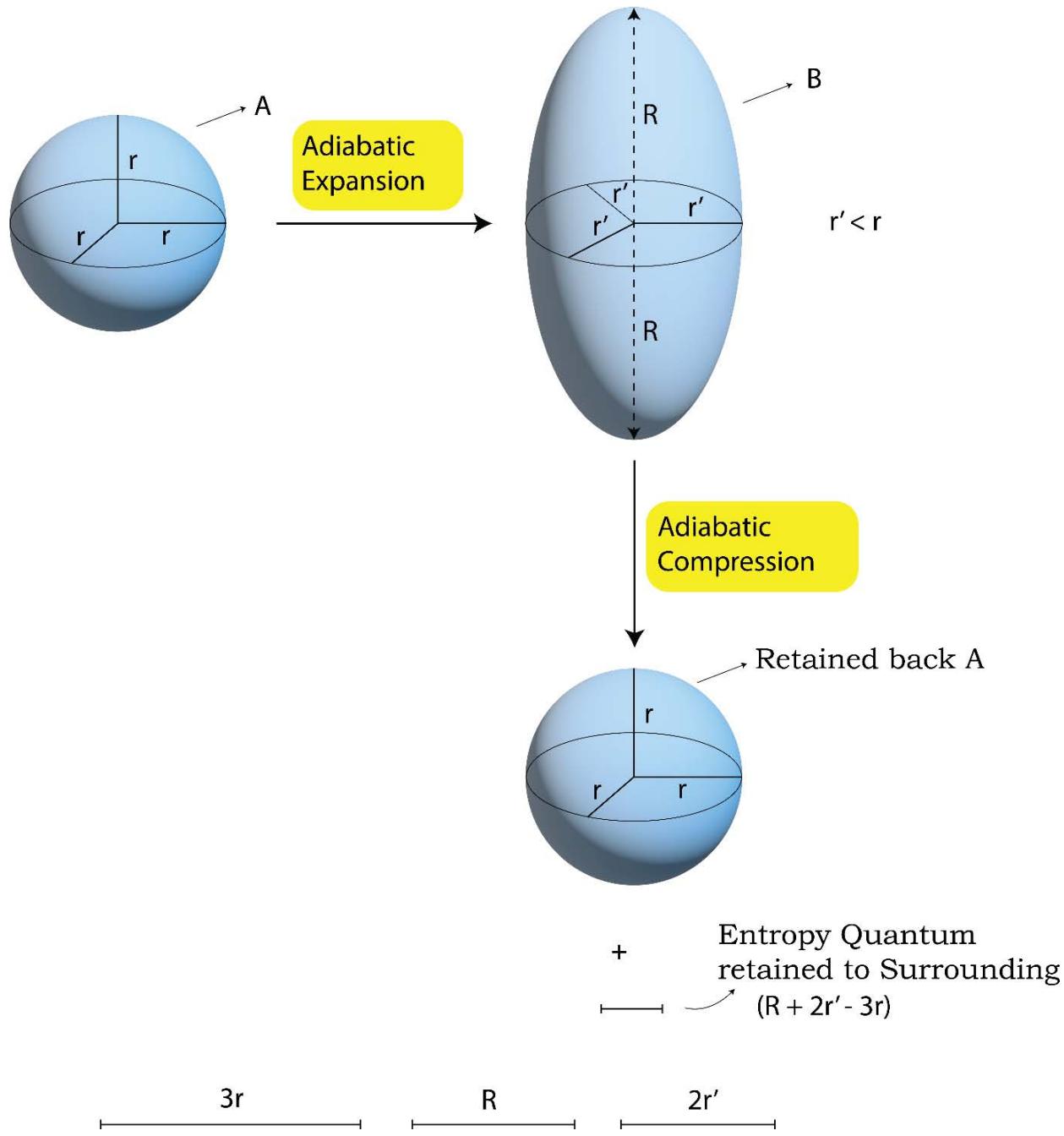 Figure 12: Quantum Level Presentation of a Typical 'Adiabatic Expansion- Compression' Process
From Figure 11 a and 11b, one would find that during an isothermal expansion the 3D volume (or energy) sphere quantum increases in its size homogeneously over all the directions in space by taking heat from the surroundings. The heat enters the system in the hybrid form of 'quantum of forces' and 'quantum of multidirectional entropies' (since heat or energy in its integrated form is the product or hybrid of force and multi directional entropy). The initial entropy is the entropy of the system only and is equal to the sum of the radius's of the sphere (over the X, Y & Z directions), $(\mathsf{r} + \mathsf{r} + \mathsf{r}) = 3\mathsf{r}$. Since during the expansion, the system encroaches some space of the surroundings, the entropy of the final state or the expanded state has to be considered in the form of the entropy of the 'system + surroundings' and would be equal to the sum of the radius's of the expanded sphere (over the X, Y & Z directions), $(\mathsf{R} + \mathsf{R} + \mathsf{R}) = 3\mathsf{R}$ $(\mathsf{R} > \mathsf{r})$. The contribution of the surroundings to the total entropy of the 'system + surroundings' of the expanded state, is 3 $(\mathsf{R} - \mathsf{r})$ such that the total entropy of the final state involving both the system and the surroundings is,
Total entropy of the final state of the 'system + surroundings'
- = (entropy of the system) + (entropy of the surroundings)
- $= (3\mathsf{r}) + (3\mathsf{R} - 3\mathsf{r})$
- $= 3R$ (2.11)
Since $R > r$, in the isothermal expansion processes the multi directional entropy is generated in the 'system + surroundings. When the isothermal compression takes place, the generated multi directional entropy is being returned to the surroundings homogeneously over all the directions in the space. So the heat is being returned back to the surroundings in the form of quantum of forces and quantum of multi directional entropies as shown in Figure 11b (in the form of a smaller 3D yellow color sphere). However, the multi directional entropies are being dissipated over all the directions in space.
In case of an adiabatic expansion-compression process as shown in Figure 12, the scenario is different than an isothermal expansion process. In this case during the expansion no heat enters the system (its own internal energy is used up) but the system while expanding, a part of the multi directional entropy of the system transforms into the directional entropy and passes on to the surroundings and some entropies of the surroundings are also being captured. It is often told that, an adiabatic process is the one in which no heat or energy exchange takes place with the surroundings. While this dictum is being true but however, is not being a complete one. In an adiabatic process, there happens an exchanges of 'entropy' quantum. This is to be noted very meaningfully that for the mechanical expansion or contraction processes as are being discussed here, there are the movements of the piston. So either the piston encroaches the distances of the surroundings while expanding or it leaves the distances to the surroundings while contracting. It is indeed the physical variable 'distance', which in fact represent the 'entropies' of the universe and hence the exchanges of entropies are always being there irrespective of the nature of the process be it isothermal, adiabatic or the others. The adiabatic processes are 'iso-entropy' in regard to the system but when the system and surroundings are being considered together, the exchange of entropies take place as is being shown below.
As shown in Figure 13, the system expands directionally (either longitudinal or lateral fashion along one of the 3 principal axes's of the space) such that the entropy of the 'system + surroundings' is increased.
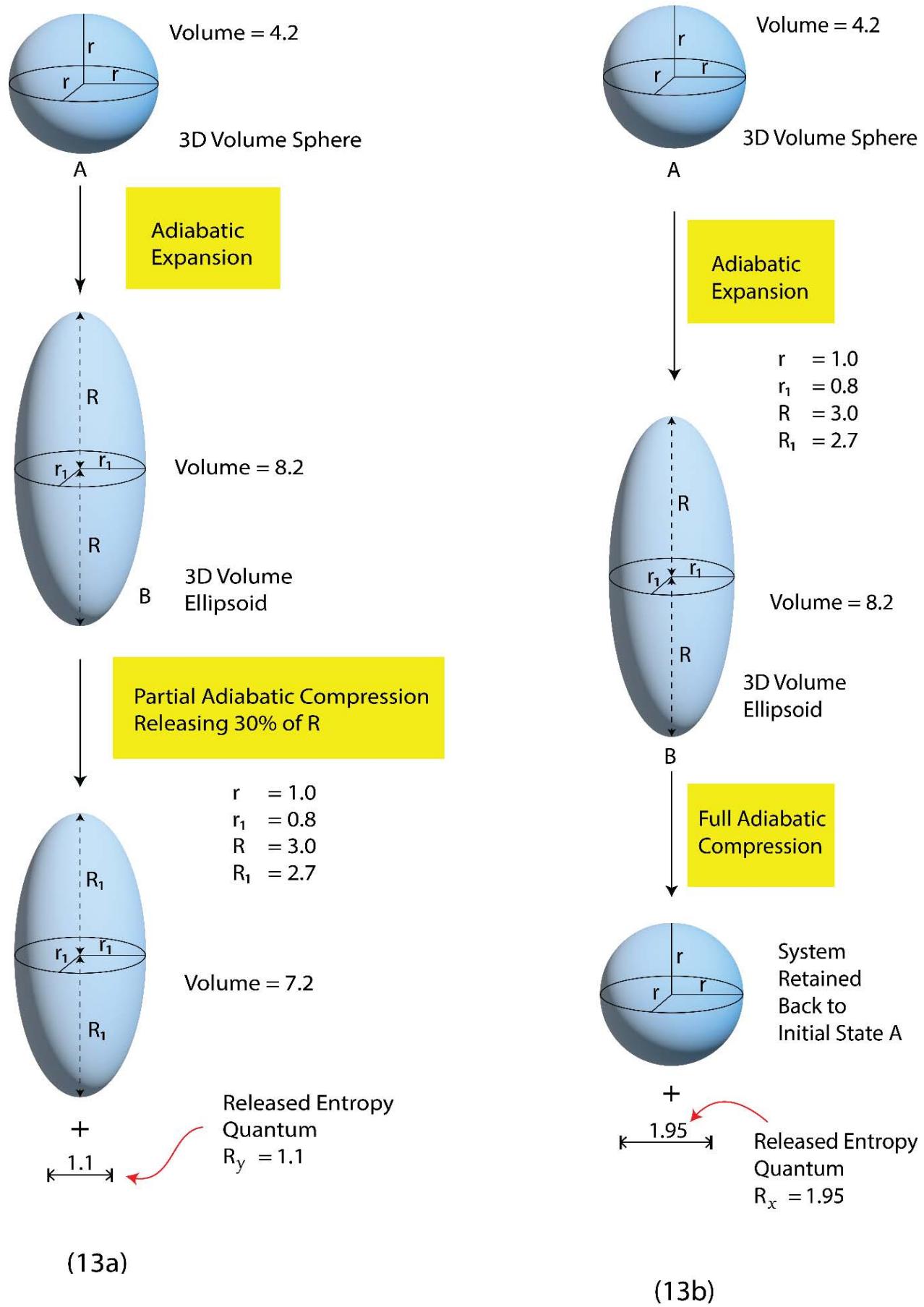 Figure 13: Quantum Level Presentation of 'Adiabatic Expansion - Full Compression' vis-à-vis 'Adiabatic Expansion - Partial Compression'
Following Figure 13a (as given below), the entropy of the system at the initial state
$$
=(r+r+r)=3r
$$
Following Figure 13a, the entropy of the 'surroundings' in the final expanded state $= R(D)$
(D to mean directional)
Following Figure 13a, the entropy of the 'system + surroundings' in the final expanded state
$$
= [3\mathrm{r} + 3\mathrm{R}(\mathrm{D})]
$$
During an adiabatic compression process since there is no question of the release of heat to the surroundings by the system, what happens is, the system returns the directional entropy quantum,3 R(D), to the surroundings as shown in Figure 13c.
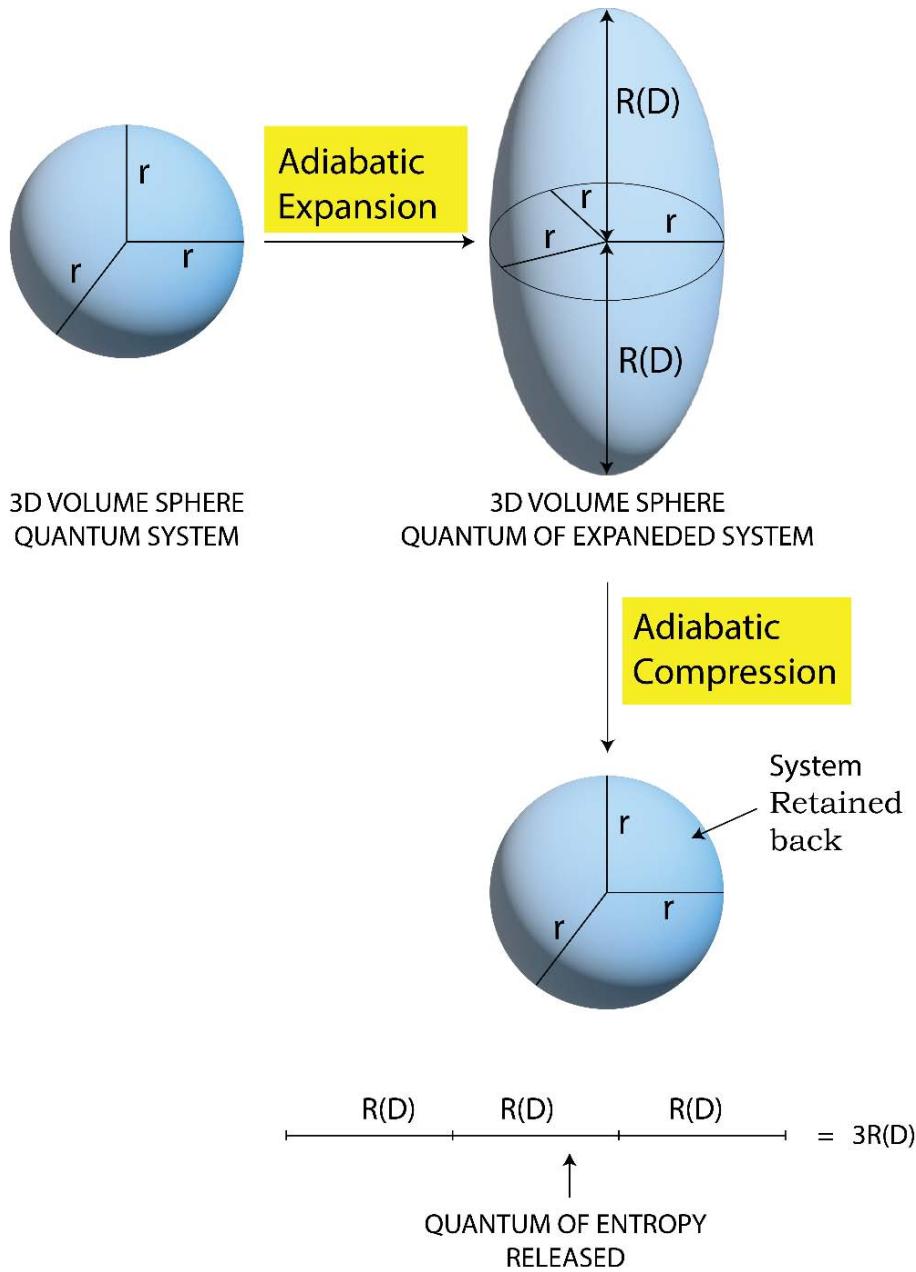 Figure 13c: Presentation of 'Directional Entropy Quantum' Generation in the Adiabatic Processes
Before proceeding further, it is very much required to clarify one 'time long misconception' which is prevailing in the subject of chemical thermodynamics and in the mind of the scientific community, from the beginning of its inception. The said misconception is regarding the conservation of the 'entropy' of an adiabatic expansion process. It has been taught that the adiabatic expansion processes are 'iso-entropy' by their nature. However, this is a wrong conception. The true conception is, in an adiabatic expansion process, the
'randomness or the energy involved' of the system in the initial state remains to be the same as that of the 'randomness or the energy involved' of the 'system + surroundings' of the final state or the expanded state. The measure of randomness of a system is 'TS' and which is the product of the temperature (T) and entropy of the 'system + surroundings' (S). Now during an adiabatic expansion process the T drops and the S increases and the said decrease and increase of T and S are very much proportionate to each other such that the product of the two remains to be at the same level before and after the commencement of an adiabatic expansion process. In case of an isothermal expansion process, the T remains to be constant and S increases from the initial state to the final state and as result of that both the 'entropy' and the 'randomness' (TS) increases.
For an adiabatic expansion process if $S_{1}$ and $S_{2}$ be the entropies of the initial and the final states and $T_{1}$ and $T_{2}$ being the temperatures of the initial and final states respectively and since no heat does enter or leaves the system, one can write,
$$
T _ {1} S _ {1} = T _ {2} S _ {2} \tag {2.13}
$$
The above said equivalence of T and S is evolved from the fact that the involvement of 'energy' between the initial state and the final state are the same since no heat enters or leaves the system and the composite parameter 'TS' is the representation of the 'involved energy' of a thermodynamic state.
The above said concept of TSQ is quite different from the conventional concept. The conventional concept labels an adiabatic process as an 'iso entropic' one and hence $S_{1}$ is being put equal to $S_{2}$. The readers of this research article are being requested to please come out of this non-appropriate approach mainly on ground of the following important facts:
1. 'S' represents the entropy of the 'system + surroundings' in the famous Helmholtz's equation of free energy (in the form $F = U - TS$ ).
2. $S_{1}$ represents, the entropy of the 'system + surroundings' at the initial state but the entropy of the surroundings is zero since the system has not started to encroach the space of the 'surroundings'.
3. In the final state while the entropy $S_{1}$ of the system is being retained, but a positive contribution of the surrounding's entropy is added to the system entropy (since the surroundings is being encroached due to the increase in volume of the system). So $S_{2}$ becomes higher in magnitude than the magnitude of $S_{1}$.
4. The temperature of system drops since solely the internal energy of the system is being utilized in the work done of the expansion and as result $T_{2}$ turns out to be lesser than $T_{1}$.
The magnitude of the product of 'higher temperature-lower entropy' of the initial state counter balances the said magnitude of 'lower temperature - higher entropy' of the final state such that $T_{1} S_{1}$ becomes equal to $T_{2} S_{2}$ or $T_{1} S_{1} = T_{2} S_{2}$.
The one of the failures of the conventional thermodynamics lies in its non-ability to calculate the free energy change of an adiabatic expansion or compression process. For an adiabatic expansion (in the conventional thermodynamics) $S_{1}$ is being put equal to $S_{2}$ and is being represented as 'S' for both the initial and the final states. Now, if $F_{1}$ and $F_{2}$ and $U_{1}$ and $U_{2}$ are being the free energies and internal energies respectively of the initial and final states, then as per conventional thermodynamics,
$$
F _ {1} = U _ {1} - T _ {1} S
$$
$$
F _ {2} = U _ {2} - T _ {2} S
$$
So, the change in free energy,
$$
\Delta F = F _ {2} - F _ {1} = \left(U _ {2} - U _ {1}\right) - S \left(T _ {2} - T _ {1}\right) \tag {2.14}
$$
$$
= \Delta U - S \Delta T
$$
From the above expression (2.15) it is not at all being possible to calculate $\Delta F$, since it requires to know the absolute value of the 'entropy' of a system and which is however not possible to evaluate in conventional thermodynamics. This is the reason; this topic has been avoided in the conventional subject of chemical thermodynamics.
The true expression for the change in free energy based on the above discussions on 'adiabatic process' is,
$$
F _ {1} = U _ {1} - T _ {1} S _ {1}
$$
$$
F _ {2} = U _ {2} - T _ {2} S _ {2}
$$
So, the change in free energy,
$$
\begin{array}{l} \Delta F = F _ {2} - F _ {1} = (U 2 - U _ {1}) - (T _ {2} S _ {2} - T _ {1} S _ {1}) \\= \Delta U [ \text{since}, \left(\mathrm{T} _ {2} \mathrm{S} _ {2} - \mathrm{T} _ {1} \mathrm{S} _ {1}\right) = 0 ] \tag{2.16} \\\end{array}
$$
Now according to the $1^{\text{st}}$ law of thermodynamics, $\Delta Q = \Delta U + \Delta W$ ( $\Delta Q$ and $\Delta W$, are heat absorbed and the work done respectively) For an adiabatic process since $\Delta Q = 0$
$$
\Delta F = \Delta U = - \Delta W
$$
$$
Or\\Delta F = - \Delta W
$$
$$
Or\\Delta\mathrm{U} = -\Delta\mathrm{W}
$$
So for the adiabatic processes under discussions, a part of the internal energy of the system $(\Delta U)$ is being fully converted to work. In fact within the system for the adiabatic processes, $\Delta U$ bears the same significance as that of $\Delta Q$ for the non-adiabatic processes. The full conversion of energy (or heat) to work does happen in the adiabatic processes. The second law of thermodynamics is not applicable for the adiabatic processes.
For an isothermal process, with the same initial and final states as that of the adiabatic process as just discussed above, the expression of $\Delta F$ will be,
$$
\Delta F = \Delta U - \left(T _ {2} S _ {2} - T _ {1} S _ {1}\right) \tag {2.18}
$$
But since for an isothermal process the temperature remains constant, one can put, $(T_{2} = T_{1} = T)$ and since $U = 0$ too, the above expression reduces to,
$$
\Delta F = T \left(S _ {2} - S _ {1}\right) = T \Delta S \tag {2.19}
$$
Now in the equation, $\Delta Q = \Delta U + \Delta W$ if $\Delta Q$ is put equal to $\Delta S$ [since as far as the conventional definition of $\Delta S$ is concerned, it is ( $\Delta S = \Delta Q / T$ )] and since $\Delta U = 0$, the said equation of $\Delta Q$ becomes,
$$
\Delta Q = T \Delta S = \Delta W
$$
The temperature of initial state $A = (4\pi r^2 / 3) = T_1$
The temperature of final state $\mathsf{B} = (4\pi r_1^2 / 3) = T_2$
The entropy of the initial state $\mathsf{A} = (\mathsf{r} + \mathsf{r} + \mathsf{r}) = 3\mathsf{r} = \mathsf{S}_1$
The entropy of the final state $\mathsf{B} = (\mathsf{r}_1 + \mathsf{r}_1 + \mathsf{R}) = (2\mathsf{r}_1 + \mathsf{R}) = \mathsf{S}_2$
Now for the adiabatic process as discussed,
$$
\mathrm {T} _ {1} \mathrm {S} _ {1} = \mathrm {T} _ {2} \mathrm {S} _ {2}
$$
So, $(4\pi r^2 /3)\times 3r = (4\pi r_1^2 /3)\times (2r_1 + R)$
$$
Or, 3r^3 = r_1^2 (2r_1 + R)
$$
Now another point to note that since the temperature is dropping in an adiabatic expansion process and considering the mathematical expressions of $T_{1}$ and $T_{2}$ as shown above it can be concluded that
$$
(4 \pi r ^ {2} / 3) > (4 \pi r _ {1} ^ {2} / 3)
$$
So $r > r_1$ (2.22)
So the lateral radius $(r_1)$ of the ellipsoid has to be smaller in magnitude to that of the radius of the temperature circle of the initial state and which is being $r$. So, the force circle of the initial state will be laterally But in reality $\Delta Q$ remains always higher than $T\Delta S$ for the isothermal processes (will be discussed in detail in next sections to follow) and hence $\Delta Q$ also remains higher than $\Delta W$ for the very obvious reasons there off. Hence heat cannot be completely converted to work in practice for the isothermal processes.
Before proceeding further, the physics, the geometric profile and the mathematics of an adiabatic process needs to be understood very well. In Figure 13, an adiabatic 'expansion - compression' topology of a system (expansion from the initial state A to final state B and compression from final state B to initial state) has been presented schematically.
In the volume sphere A as shown in Figure 13, a force circle is residing of radius equal to r and as per TSQ, this radius is related to the temperature of the initial state. The volume sphere A has been shown to expanded arbitrarily by a magnitude of $50\%$ and unlike an isothermal expansion process the final state B has taken an ellipsoid shape (instead of a spherical shape in case of an isothermal expansion) of 'longitudinal radius' being equal to R and the two numbers of 'lateral radius' both being $\mathsf{r}_1$. The underlying logic behind the occurrence of attainment of the ellipsoid shape has already been discussed. The temperature of the final state would be related to the radius of the base circle $(\mathsf{r}_1)$ of the ellipsoid B. The following mathematics of the adiabatic process needs to be followed now,
Now it will be shown that how the 'space-time'[1-8] of the universe is related to the following two very to very important thermodynamic phenomena
1) In an isothermal expansion process $\mathrm{d}Q > \mathrm{TdS}$ such that the heat cannot be completely converted to work.
2) In an adiabatic process, $\Delta U = -\Delta W$ such that a part of the internal energy of the system is fully converted to work.
The 'space-time' of the universe as shown in Figure 4, is embedded in an atom of the baryonic matters in the form of [(energy x entropy x time)] or [(space x entropy x time)] or [(volume x entropy x time)].
In TSQ, the 'space', 'energy' and 'volume' are dimensionally are the same.
In an 'energy/volume' quantum, the 'entropy' and 'time' quantum are both existing. As shown in Figure 15, the 'heat or energy' 3D quantum(P) is representing the dQ. This dQ quantum is holding time 2D-saddle quantum and the unidimensional entropy quantum (S). Now when this 'dQ' passes isothermally on to its differential form from its integrated state in the form of force circle(T) and distance, dS (R), what is evolved is the well known 'thermodynamic work' in the form of (Force x distance) and is clearly shown in Figure 14.
#### Case of dQ = Tds Full Conversion of Heat into Work (Ideal gas)
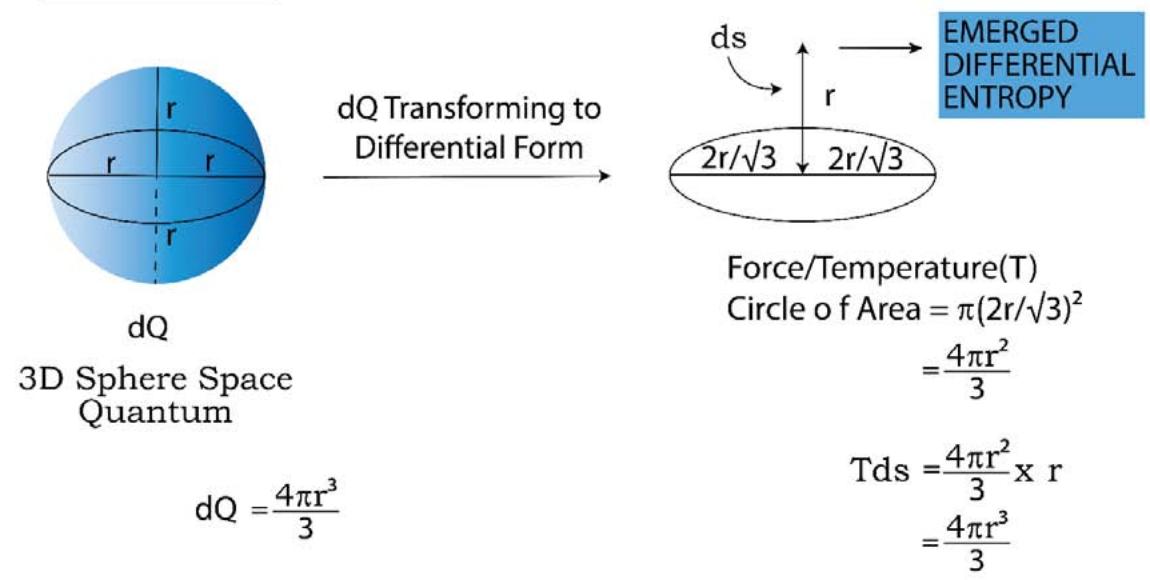
Figure 5 above) and the Break Up of the 'Energy Quantum' to 'Internal Energy Quantum, U' (superposition of 2 numbers of volume Quantum) and 'Volume Energy Quantum, PV'
#### Case of dQ > Tds Partial Conversion of Heat into Work (Real system)
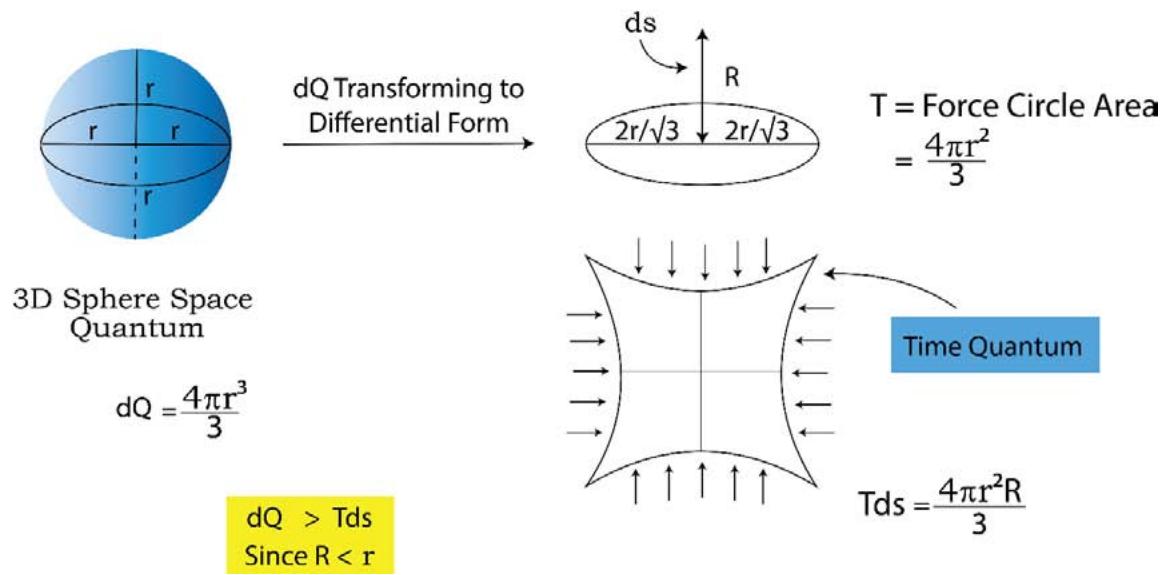 Figure 14: Presentation of Co- relation between dQ and TdS for the Ideal and Non-ideal Working Substances
The presence of the '2D time quantum' which is 'pull back quantum' (attractive force) keeps the entropy quantum at its own strong hold and does not allow the 'S' to completely emerge out from the 3D sphere. As a result of this dS (or R) turns out to be lesser than S.
$$
\begin{array}{l} \text{SoTdS} = \text{(Temperaturexchangeinentropy)} \\= \left(\text{areaofcircleT}\right) \times \left(\text{lengthR}\right) \\= T d S \\= \left[ \pi (2 r / \sqrt{3}) ^ {2} \right] \times R \\= \left[ \pi (2 r / \sqrt{3}) ^ {2} \right] \times R \tag{2.23} \\\end{array}
$$
$\left[\mathrm{d} \mathbf{S} = \text { change in entropy } = (\mathbf{R} - 0) = \mathrm{d} \mathbf{S}\right.$, since before the transformation of the dQ quantum from the integrated 3D form to the differential form, the entropy was fully latent within the 3D sphere and its contribution was zero]
The energy/volume of the sphere Por
$$
\mathrm {d} Q = (4 \pi r ^ {3} / 3) ] = [ \pi (2 r / \sqrt {3}) ^ {2} ] \times r ] \tag {2.24}
$$
If equations (2.23) and (2.24) are compared to each other, one will find that
$$
\mathrm{d Q} > \mathrm{T d S} (\text{sinceR} < \mathrm{r}) \tag{2.25}
$$
We have been taught that in an isothermal reversible expansion of an ideal gas, the heat is fully converted to work. Let this paradigm be examined in the light of TSQ. If the system would be an ideal gas instead of any real gas or others, the time graviton (the attractive physical variable) would have disappeared and the entire 'r' would have emerged out of the 3D energy/heat quantum as shown in Figure 15, and TdS would be equal to dQ (since R would be equal r as shown in Figure 14) and hence the full conversion of heat to work would have taken place. So we could have written,
$$
\mathrm{T d S} = \mathrm{d Q} _ {\text{reversible}}
$$
$$
\mathrm {d S} = \left(\mathrm {d Q} _ {\text {r e v e r s i b l e}}\right) / \mathrm {T} \tag {2.26}
$$
By the above equation (2.26), the entropy had been defined in conventional thermodynamics without putting the proper logic behind such definition of 'entropy'.
This above made discussions and argument very clearly establishes the fact that why heat cannot be completely converted into work. The conventional subject of thermodynamics depicted the said fact in the form of '2nd law of thermodynamics' but could never furnish a satisfactory explanation of the root cause of this phenomenon. Only through the consideration of 'space-time' realm of the universe and the concepts of
TSQ this is now being revealed very much clear cut indeed.
To understand the fact that in case of the adiabatic processes as discussed above, the $2^{\text{nd}}$ law of thermodynamics fails and full conversion of a part of internal energy (or heat) to 'work' occurs, one needs to study the topology of internal energy of a system as shown in Figure 15. While in case of an isothermal process, the 'PV' part (or volume energy part) of 'enthalpy, H' is involved (at the occurrence of volume expansion/contraction), in the case of adiabatic processes, for the said occurrence, only the internal energy part is being involved. From Figure 16, it is clearly found that while in the 'PV' part there does exist a single force circle, the 'internal energy part' contains two numbers of force circles. In the following figure 15, it is shown geometrically, how the said force circles squeezes and generates very larger magnitude 'directional' entropies. The uniqueness of the said happenings of the squeezing actions lies in the fact that as a result of this, the magnitudes of the 'entropy quantum' increases but the magnitude of the 'internal energy quantum' decreases. It is, however being a known fact that during the adiabatic expansion processes the internal energy does go on decreasing.
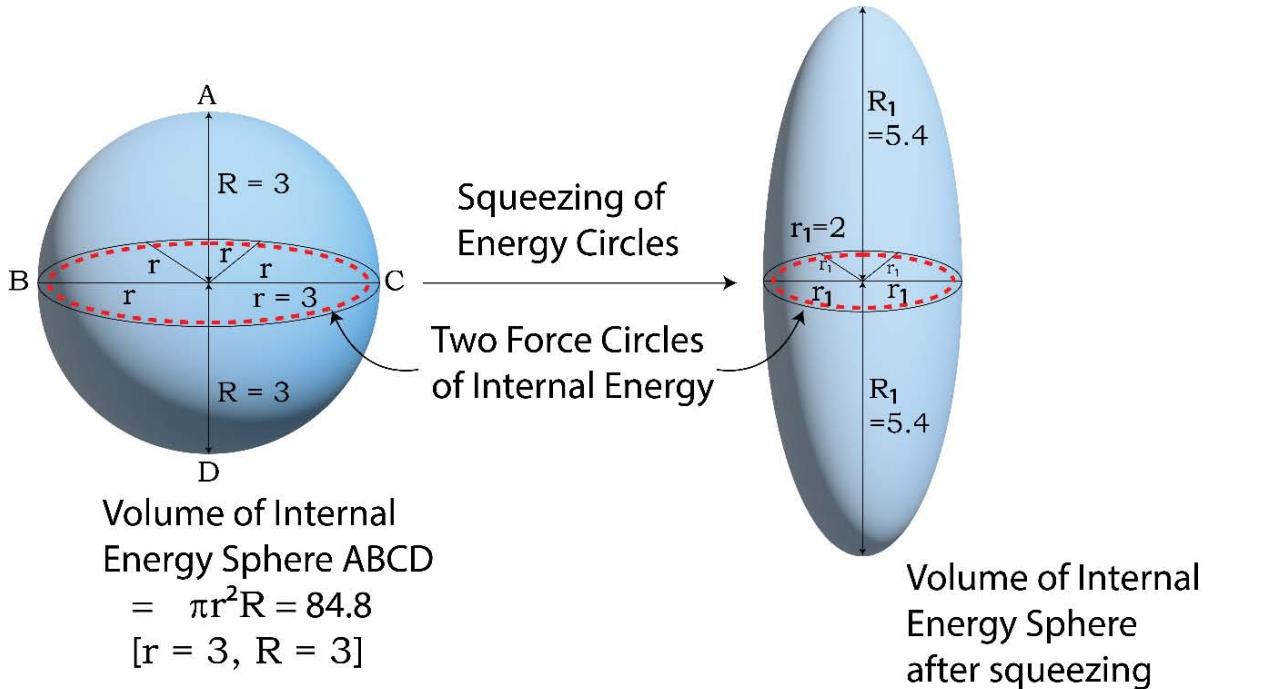 Figure 15: Squeezing of the 2 numbers of 'Internal Energy Force Circles' to Generate 'Higher Magnitude Entropy Quantum'
$$
= \pi r _ {1} ^ {2} R _ {1} = 6 7. 8
$$
$$
[ \mathrm {r} _ {1} = 2. 0, \mathrm {R} _ {1} = 5. 4 ]
$$
% Decrease in Internal Energy
$$
= \frac {\pi r ^ {2} R - \pi r _ {1} ^ {2} R _ {1}}{\pi r ^ {2} R} \times 100 = 20 \%
$$
$\%$ Decrease in Force
$$
= \frac {\pi r ^ {2} - \pi r _ {1} ^ {2}}{\pi r ^ {2}} \times 100 = 55.5 \%
$$
% Increase in Directional Entropy
$$
= \frac {\mathrm {R} _ {1} - \mathrm {R}}{\mathrm {R}} \times 100 = 80 \%
$$
From the above Figure, it is clearly revealed that while the force circles are squeezed by their areas by say $55.5\%$ (as for example), the length of the directional entropy quantum (R) increases by about $80\%$ but such occurrence leads to an overall diminution in internal energy by about $20\%$.
Since in the cases of the adiabatic processes, the two force circles are working in tandem with each other to generate the 'directional entropy', a very high dispersive or 'push forward' state of affair is arisen out and as a result of that, the said high 'push forward force' fully overcomes the 'pull back time quantum' and the directional entropies(unlike the case of isothermal process) commensurate to its required magnitude for the full conversion of heat to work, may emerge out of the said 'time attractive cage'. As a result of that the full conversion of 'heat' to 'work' takes place in the concerned adiabatic processes.
## III. THE FUNCTIONS OF A HEAT ENGINE OF A TYPICAL 'CARNOT ENGINE'
1) At the very beginning the system takes up a certain quantity of heat from a high temperature 'source' in the form of 'force quantum' and 'multi directional entropy quantum' and a thermodynamic equilibrium is attained between the source and the system and the system acquires the same temperature as that
of the source. Let the absorbed heat be $Q_{1}$ by its magnitude.
2) Then in the $1^{\text{st}}$ step of the Carnot cycle, which is an isothermal expansion, another certain quantity of heat enters the system and which is fully utilized in the mechanical expansion. Let the heat be $Q_{2}$ by its magnitude. The volume of the system increases from $V_{1}$ to $V_{2}$
3) So, the total heat input in the system is $(\mathsf{Q}_1 + \mathsf{Q}_2)$ and it can be expressed in the form of 'force' and 'multi directional entropy' as, $(\mathsf{Q}_1 + \mathsf{Q}_2) = (\mathsf{F} \times \mathsf{X})$ where $\mathsf{F}$ stands for the magnitude of force and $\mathsf{X}$ stands for the magnitude of multi directional entropy.
4) The second step of the Carnot cycle is an adiabatic expansion process and let, out of the total multi directional directional entropy 'X', an amount of entropy 'Y' is transferred to the directional entropy. So multi directional entropy of amount (X -Y) is retained back. The volume of the system increases from $V_{2}$ to $V_{3}$.
5) The $3^{\text{rd}}$ step of the Carnot cycle is an isothermal compression and it releases heat to the surroundings in the form of the product of amount of 'force, F' and the amount of multi directional entropy (X-Y), as [F x (X-Y)]. The multi directional entropy of amount (X-Y) is being dissipated all over the surroundings. The volume of the system decreases from $V_{3}$ to $V_{4}$.
6) In the $4^{\text{th}}$ step of the Carnot cycle, which is an adiabatic compression step, the 'directional entropy,' $\Upsilon'$ is being released to the surroundings and the system returns back to the initial state. The volume of the system decreases from $\mathsf{V}_4$ to the original volume of the system $\mathsf{V}_1$.
7) The 'quantum of force' which is released in the step 3 and the 'quantum of directional entropy,' 'Y' which is released in the step 4, combines with each other and forms a 'quantum of differential heat, FY' and which is the 'thermodynamic work output' of a typical Carnot cycle.
The 'topology' of a typical Carnot cycle as described above is shown in Figure 16 below:
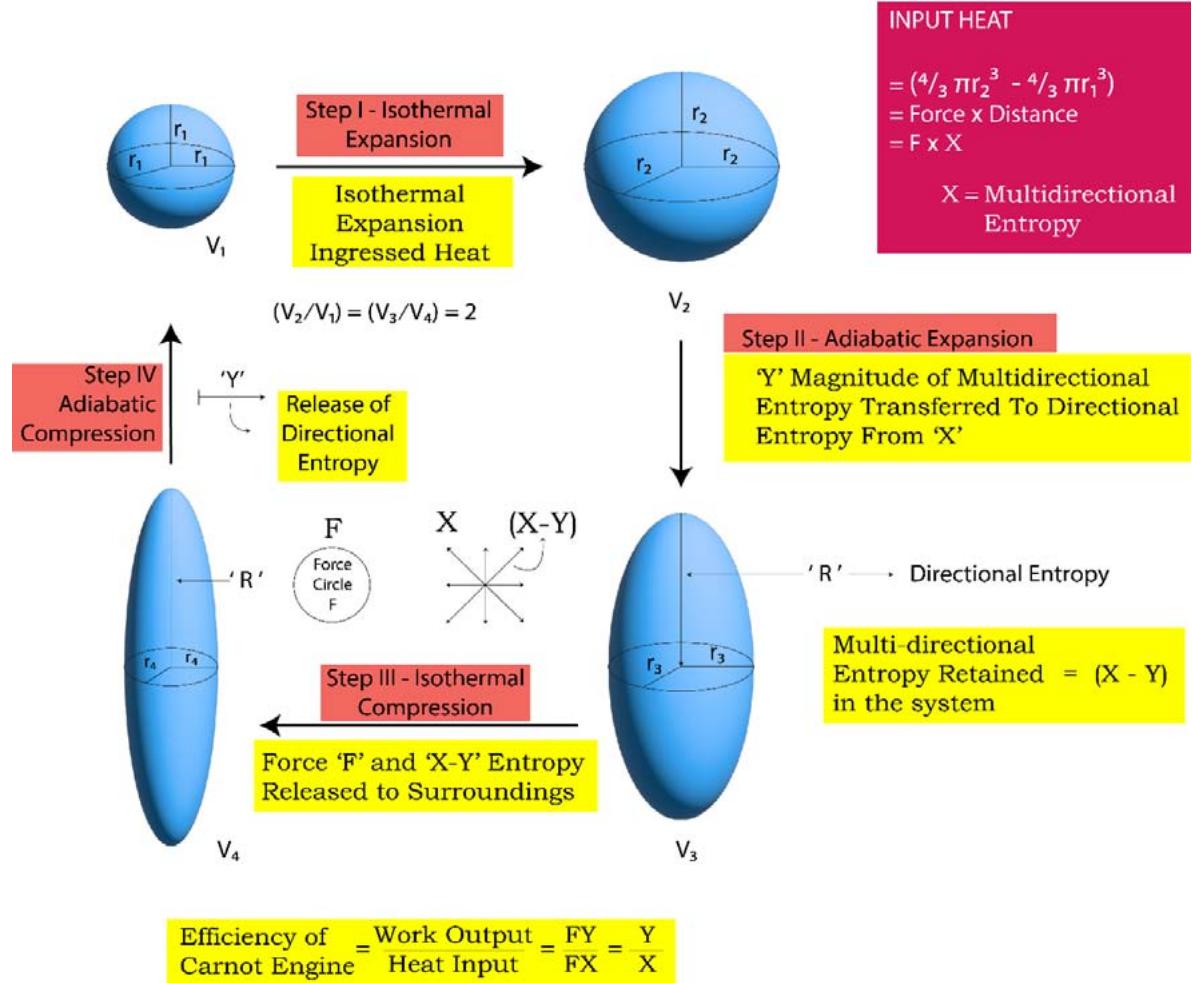
TOPOLOGY OF A TYPICAL CARNOT ENGINE - 3D PRESENTATION Figure 16: Topology of a Typical Carnot Heat Engine to Generate 'Thermodynamic Work' For many to many years people go on calculating the efficiency of a heat engines in the following way:
Efficiency of a heat engine = (Net work done by the engine in the different steps in a complete cycle/ Input heat)
This is notably being unjustified since what is to be evaluated is, how much of 'thermodynamic work' output a heat engine is generating (and which passes out to the surroundings) against the input heat. The corrected or true mathematical formula for calculating the efficiency is:
The efficiency of a typical Carnot engine =
$$
(Thermodynamic work output / heat input) = (FY /FX) = (Y/X) =
$$
(magnitude of directional entropy which is created in the adiabatic expansion)/(magnitude of multi directional entropy which is introduced in the system in the isothermal expansion at the beginning) (3.1)
Since Y is always being less than X, the efficiency of a Carnot engine is always less than 1 or unity. The working substance of a typical Carnot engine is an ideal gas and which is maximum stretchable such that the directional entropy 'Y' can attain its maximum value (though 'Y' always remains to be less than 'X') in the adiabatic expansion step and hence it is being concluded that 'the efficiency of a typical Carnot engine is maximum'.
Indeed, this is to mention here that due the lack of the proper notion of what the 'efficiency' of a heat engine is, a wrong conclusion had been made at past that 'the efficiency of a Carnot heat engine is independent of the nature of the working substance' and this dictum is still persisting. As a matter of fact, while compared among multiple working substances, the one which is most stretchable will yield the highest value of 'Y' and hence the efficiency of the same will be the highest for that particular working substance among the all. The converse of this statement is also being true.
While a typical Carnot engine is being a 4-stroke engine (two numbers forward strokes of expansions, the isothermal and the adiabatic ones and another two numbers of backward strokes of compressions, the isothermal and the adiabatic ones too), a question might arise that why 2-stroke back and forth heat engines are not being fabricated with the arrangements like 'an isothermal expansion' followed by an isothermal compression' or 'an adiabatic expansion' followed by an 'adiabatic compression' since in both the said arrangements the back and the forth motions of the piston of the heat engine are happening. This is being explained below:
i) Regarding a hypothetical 2-stroke 'isothermal' expansion-compression heat engine, this is to note that the temperature of the working substance in an isothermal expansion process remains to be very high and which is same to same as that of the temperature of the high temperature 'source'. Under this situation when the system is being put to the low temperature 'sink', it takes pretty long time to compress back to the lower temperature. In a heat engine what is required utmost are the very fast forward and backward movements of the piston and in this case though the forward movement of the piston would be pretty fast but since the backward movement of the piston would be quite slow, such engines will not be a workable solution in practice. While this argument is being a very tangible one from the angle of the practical operability of the engine, the argument which is being very fundamental one is, an isothermal compression process liberates heat in the 'integrated' state in the 'hybrid' form of the quantum of 'force' and quantum of 'multi directional entropy' but what is required as an output from a heat engine is, the release of heat in the 'differential' or the 'dehybridized' state in the form of quantum of 'force' and quantum of 'directional entropy' and it is the 'directional entropy only, which drives a vehicle towards a 'definite direction' and the above said 'dehybridized' or the 'differential 'state of heat is indeed what a 'thermodynamic work' is. If the output heat from a heat engine would be an integrated one in the hybrid form of 'quantum of force' and 'quantum of multi directional entropy', the wheels of a vehicle would go on rotating and rotating at the same place and could neither move forward nor could it move backward or cover distances.
ii) Regarding a hypothetical 2-stroke 'adiabatic' expansion-compression heat engine, this is to note that the system is an isolated one and no heat exchange can take place with the surroundings. So the forward and backward motions of the piston cannot be initiated. Also to note, while compressing the system releases only the quantum of 'directional entropy' but no 'force quantum' is made available to get associated with the said 'directional entropy' and hence no 'thermodynamic work' could be generated.
In the derivation of the free energy equations of G and F in the previous section, it has been firmly established that the entropy parameter 'S' in the said equations represent the 'entropy of the system + surroundings' and hence the 'T-S' or 'temperature-entropy' diagrams of a typical Carnot engine needs to be reconstructed. The conventional 'T-S' diagram of the 'conventional chemical thermodynamics' vis-a-vis the
TSQ driven 'T-S' diagram for a typical Carnot engine are shown below in Figure 17:
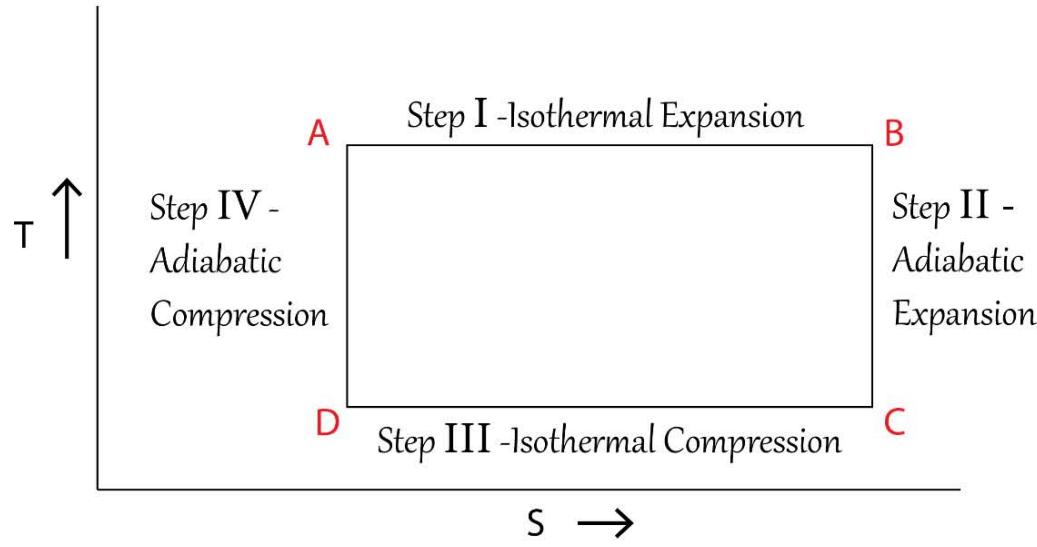
Temperature(T) - Entropy (S) Diagram of a Typical Carnot Cycle -As per Conventional Thermodynamics
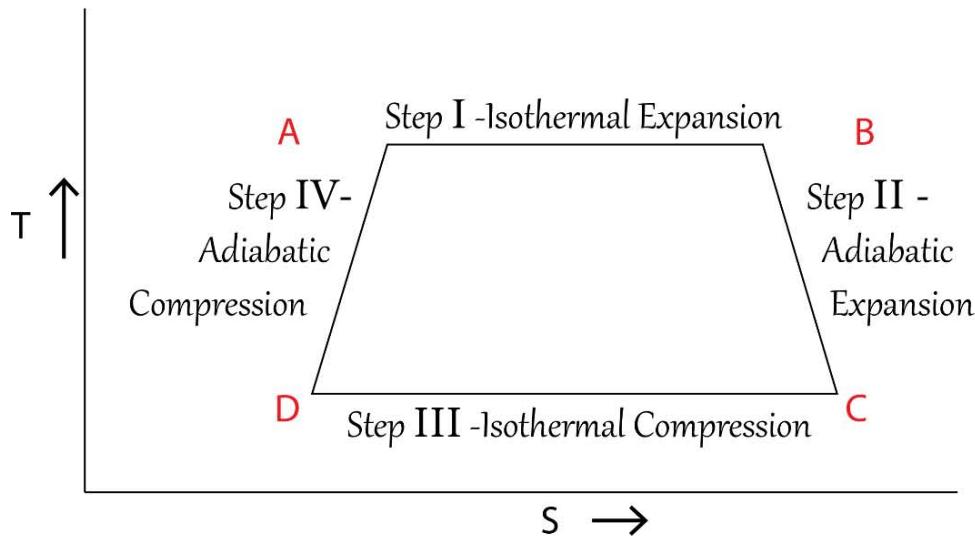
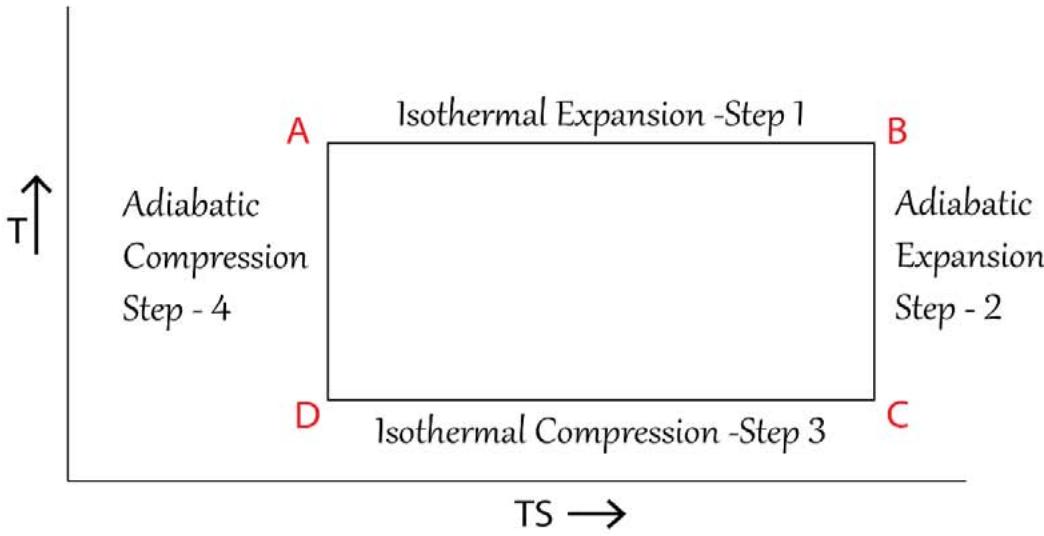
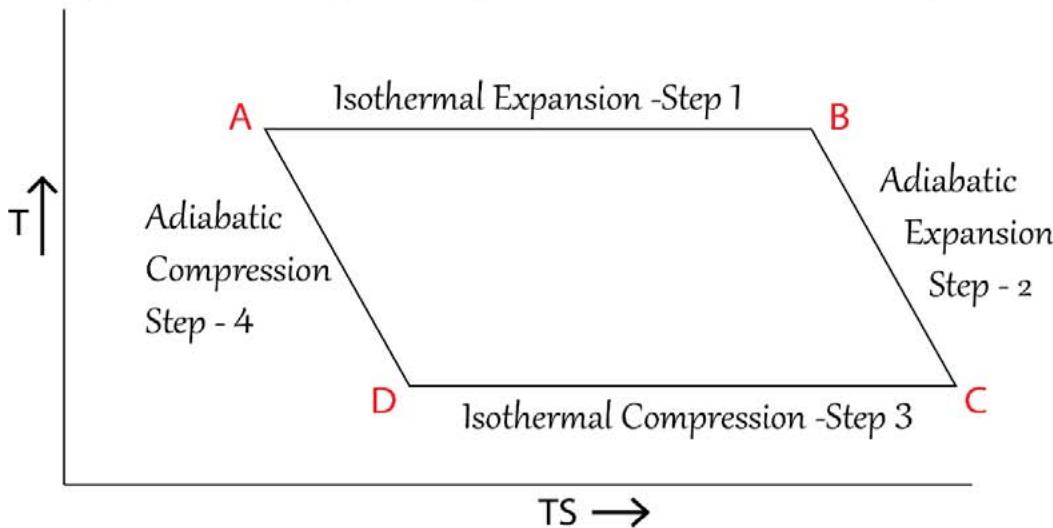
Temperature(T) - Entropy (S) Diagram of a Typical Carnot Cycle -As per TSQ Thermodynamics Figure 17: 'Temperature (T) - Entropy (S)' Diagram of a Typical Carnot Cycle as per TSQ and the Conventional Thermodynamics From the above Figure 17, it is very clearly found that while in the conventional diagram in view of erroneously considering S as the entropy of the system only, the area under 'T-S' is a rectangle but for the very obvious and the valid logic as discussed, the TSQ predicted 'T-S' diagram is a rhombus type since the entropy S (of the system+ surroundings', increases to some extent in the adiabatic expansion step and decreases in the symmetric fashion in the adiabatic compression state.
If the 'T -S' diagram of the conventional thermodynamics for the Carnot cycle as shown in Figure17 is being studied closely, it is revealed that for the pair of points or the pair of thermodynamic states 'A&D' and 'B & C', the magnitudes of the entropies are being the same. From the starting point B of the $1^{\text{st}}$ adiabatic expansion process, the temperature goes on diminishing (but the entropy, S, remain constant throughout) up to the point C. The total energy involvement for the thermodynamic state B is 'T $_1$ S' and for the point C the same is $\mathsf{T}_2\mathsf{S}'$ and since $T_{1} > T_{2}$, $\mathsf{T}_1\mathsf{S}'$ turns out to be higher than $\mathsf{T}_2\mathsf{S}'$ and which means the total energy involvement of B is higher than that C. Since the path length BC is an adiabatic path length and no energy enters or leaves the system $\mathsf{T}_1\mathsf{S}'$ has to be equal to $\mathsf{T}_2\mathsf{S}'$, otherwise which the law of conservation of energy would be violated. The TSQ depicted 'T-S' diagram as shown in the same Figure 17, is justified since it is drawn based on the logic that the composite physical variable 'TS' are being the same for the pair of points 'A&D' and 'B & C'.
Conceptually what is required to be understood very much in regard to a typical Carnot Engine is, how the randomness (total energy involvement) of the 'system + surroundings' are changing over the 4 different cycles. The measure of the randomness is the composite of T and S and which is 'TS' and hence one should plot T versus 'TS' rather than T versus S as done in 'conventional chemical thermodynamics'. The T versus 'TS' plot is shown in Figure 18 below and the area under the T and TS turns out to be a rectangle indeed. For the same logic as given in the previous paragraph for the conventional 'T' versus 'S' diagram, the conventional 'T' versus 'TS' diagram is not acceptable on the ground of the violation of the law of conservation of energy.
Temperature(T) - Temperature x Entropy (TS) Diagram of a Typical Carnot Cycle -As per TSQ Thermodynamics
Temperature(T) - Temperature x Entropy (TS) Diagram of a Typical Carnot Cycle -As per Conventional Thermodynamics Figure 18: 'Temperature' (T) versus 'Temperature - Entropy' (TS) Diagram of a Typical Carnot Cycle as per TSQ and the Conventional Thermodynamics While in the isothermal expansion and compression steps, the magnitude of TS increases and decreases respectively (and the increase and decrease of the magnitude TS remains to be same), in the adiabatic expansion and compression steps, the magnitude of TS remain stagnant but the magnitude of TS remains to be higher in case of the adiabatic expansion compared to the adiabatic compression. These are explained below more explicitly:
i) In the $1^{\text{st}}$ isothermal expansion step, the value of T remains higher (same temperature as that of the source) but constant and S does go on increasing up to a certain extent. In the isothermal compression step the value of T is lower than the $1^{\text{st}}$ isothermal expansion step but remain constant and the value of S does go on decreasing to the same extent as that of the increase in S of the $1^{\text{st}}$ isothermal expansion step. The 'Carnot cycle is so designed' such that the change in the value of T between the $1^{\text{st}}$ isothermal expansion and the $2^{\text{nd}}$ isothermal compression state are very much proportionate such that the,
Increase in the value of 'TS' of $1^{\text{st}}$ isothermal expansion = Decrease in the value of 'TS' of the $2^{\text{nd}}$ isothermal compression ii) In the 1st adiabatic expansion step, the value of T does go on decreasing and the value of S does go on increasing up to a certain extent respectively. In the $2^{\text{nd}}$ adiabatic compression step, the value of T does go on increasing and the value of S does go on decreasing. The 'Carnot cycle is so designed' such that the above said decrease and increase of T and S respectively in the $1^{\text{st}}$ adiabatic expansion and the increase in T and the decrease in S respectively in the $2^{\text{nd}}$ adiabatic compression are very much proportionate to each other such that the composite variable 'TS' remain to be constant in the said adiabatic steps.
iii) The actual meaning of the statement as quoted above 'Carnot cycle is so designed' is, in a typical Carnot cycle, the volumes of the system in the different steps have been set up such that
$$
\left(\mathrm {V} _ {2} / \mathrm {V} _ {1}\right) = \left(\mathrm {V} _ {3} / \mathrm {V} _ {4}\right)
$$
Or $(\mathsf{V}_3\mathsf{V}_2) = (\mathsf{V}_4\mathsf{V}_1)$ (3.2)
The proportion of increase in volume in the $1^{\text{st}}$ isothermal expansion = proportion of decrease in volume in the $2^{\text{nd}}$ isothermal compression
The proportion of increase in volume in the $1^{\text{st}}$ adiabatic expansion = proportion of decrease in volume in the $2^{\text{nd}}$ adiabatic compression
## IV. FREE ENERGY AND AVAILABLE WORK FROM A SYSTEM
Regarding the free energy parameters (be it F or G), it has been mentioned earlier that when a system is resting at an equilibrium with the surroundings, they do not carry any significance at all. Suppose the following 3 systems are resting in equilibrium at ambient temperature under normal atmospheric pressure:
1. A glass of water
2. A thin wire made of Nylon 66 polymer
3. A gas cylinder fitted with a piston
Now, if a question is being raised that, which one among the above three system would possess the highest free energy/mol? The answer would be, since all the 3 systems are in thermal equilibrium, there is no significance of this question. Until and unless the situations of non-equilibrium are generated in the systems talking about free energy is meaningless.
Now if the systems 1, 2 and 3 are being started heating under isothermal conditions, since the magnitude of the S parameters (entropy of the system + surroundings), would start increasing and the value of 'TS' will also go on increasing, (T constant and S increasing) and it will just start predominating the value of TS [in equation, $(\mathsf{F} = \mathsf{U}\cdot \mathsf{-TS})$ $TS > U$, U is constant in an isothermal process]and F will start attaining negative value and the system would start doing work. Among the 3 said systems, the system whose S value would increase at a faster rate, it will offer more negative value of F and hence would do more work.
The most interesting case to observe is, if the system 2 instead of heating is being stretched isothermally and longitudinally by applying external force (such that the applied force is fully utilized for tensile elongation), the S will increase very fast (the muti directional entropy will decrease but the directional entropy will increase very much and will exceed the later) and as a result, the F will become more and more negative and that too very fast. The Nylon polymer can be stretched 200 to 300 times (before it breaks) of their original lengths and hence such systems would attain very high negative value of 'free energy' and would yield a considerable amount of 'thermodynamic work' indeed. In Figure19, the geometrical profiles of 'multi directional heat expansion' and 'directional mechanical stretching expansion' respectively of a system under isothermal conditions have been presented. In the upper part of the above said Figure, the heat expansion has been shown. The entropy of the system increases from 3r to $4.5\mathrm{r}$. In the lower part of the said Figure, the 'mechanical stretching' expansion of the same system has been shown and the entropy rises from 3r to $(2\mathbf{r}_1 + \mathbf{R})$, $r > r_1$ (since on directional stretching of a wire, the original cross section decreases to some extent).
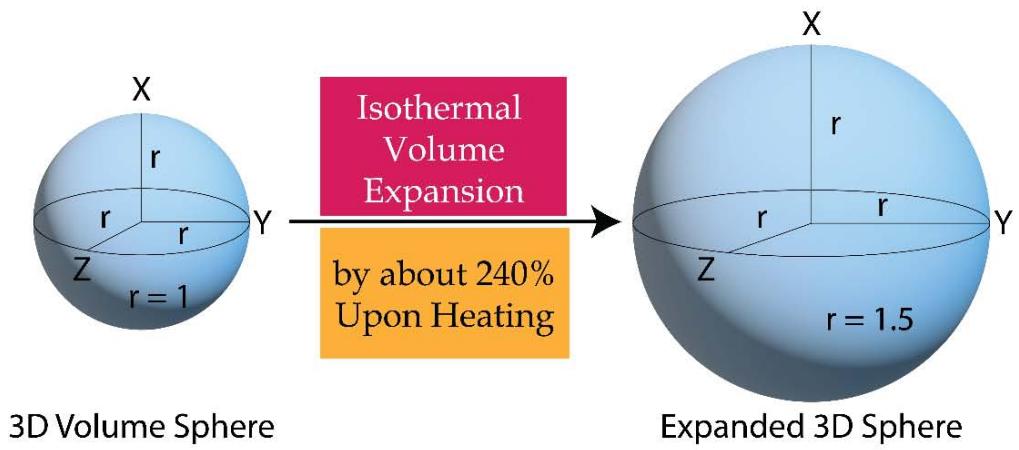
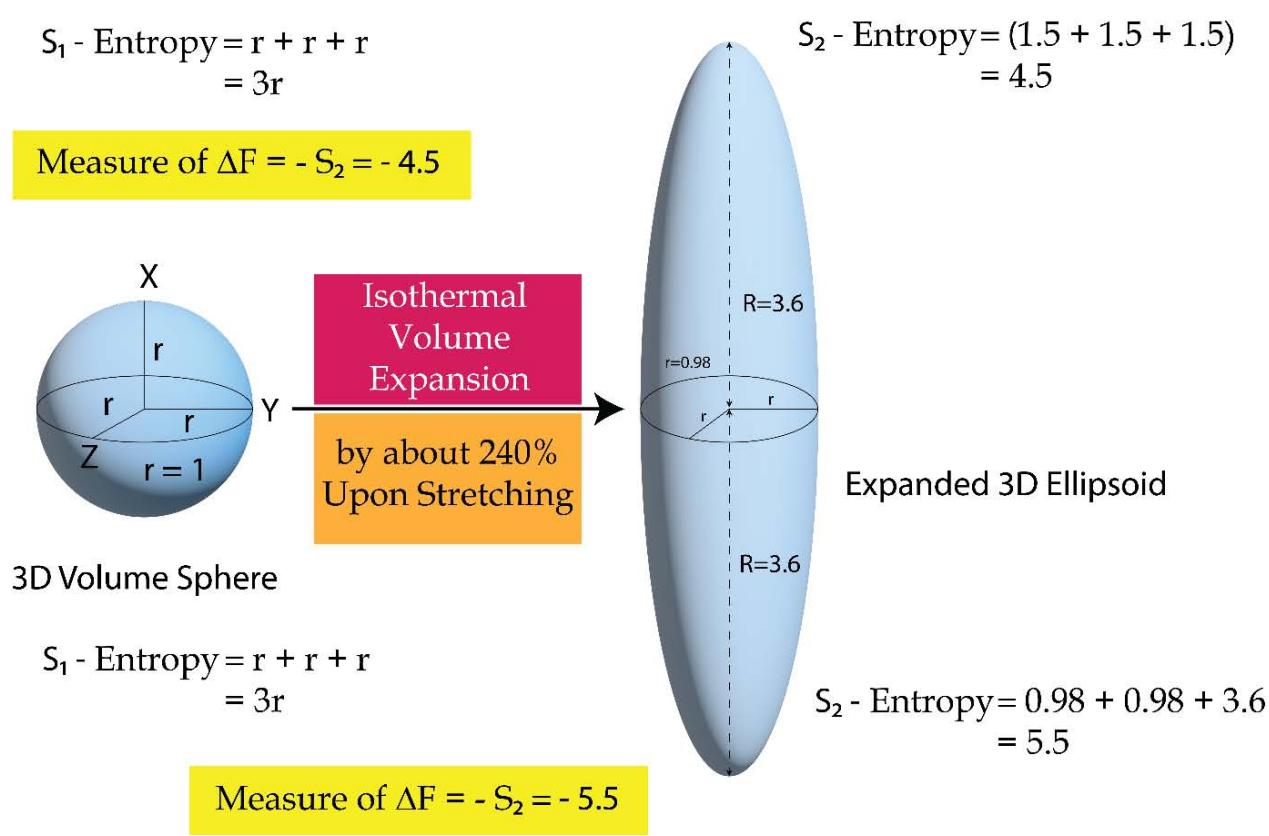 Figure 19: Geometrical Profile of a Typical 'Isothermal Volume Expansion upon Heating' vis-à-vis a typical 'Isothermal Volume Expansion upon Stretching'
In the isothermal processes, the internal energy, $U$ and the temperature, $T$ both remain constant, so, the free energy involved in an isothermal change of thermodynamic state of a system could be
If the free energy of the initial state $F_{1} = U - TS_{1}$
and the free energy of the final state $F_{2} = U - TS_{2}$
the change in free energy, $\Delta F = F_{2} - F_{1} = -T(S_{2} - S_{1})$ (4.1)
Now in the above equation for, $\Delta F$, if the cases of expansions of a same system is considered at a temperature $T$, $S_{1}$ does always remain the same and it is the value of $S_{2}$ which does change for the different levels of expansions (as for example $100\%$, $200\%$, $300\%$
respectively) but $S_{2}$ always remain higher in magnitude than $S_{1}$ and the magnitudes of $(S_{2} - S_{1})$ do always remain positive and since $T$ is constant, the measure of, $\Delta F$ could be expressed by $(-S_{2})$ only.
Table 1 below shows how the free energy $F$ of a system, attains more and more negative value (under isothermal condition) with expansion in the following fashions:
Table 1: Change in Free Energy of a System under isothermal conditions for the multi-directional and directional expansion in volume
<table><tr><td>SI no.</td><td>r0</td><td>r</td><td>R</td><td>% expansion of the volume of the system</td><td>ΔF Multidirectional (MD) S2 = -(3r)</td><td>ΔF Directional (D) S2 = -(2r0 + R)</td><td>% Higher work of D over MD</td></tr><tr><td>1.</td><td>1</td><td>1.2</td><td>2</td><td>100</td><td>-3.6</td><td>-4</td><td>5.8</td></tr><tr><td>2.</td><td>1</td><td>1.4</td><td>3</td><td>200</td><td>-4.2</td><td>-5</td><td>15.7</td></tr><tr><td>3.</td><td>1</td><td>1.6</td><td>4</td><td>300</td><td>-4.8</td><td>-6</td><td>21.4</td></tr><tr><td>4.</td><td>1</td><td>1.7</td><td>5</td><td>400</td><td>-5.1</td><td>-7</td><td>37.2</td></tr><tr><td>5.</td><td>1</td><td>1.8</td><td>6</td><td>500</td><td>-5.4</td><td>-8</td><td>48.2</td></tr><tr><td>6.</td><td>1</td><td>1.9</td><td>7</td><td>600</td><td>-5.7</td><td>-9</td><td>57.8</td></tr></table>
The graphical presentation of $\Delta F$ against the different levels of expansion is shown in Figure 20(the graphical presentation of Table 1). From the said figure one can understand at ease how the directional expansions yield higher and higher amount the work (W) compared to multidirectional expansions. In the cases of directional expansions, the values $\Delta F$ are always more negative and higher negative it be, $W$ will be higher since $W = (\Delta F)$ for an isothermal process.
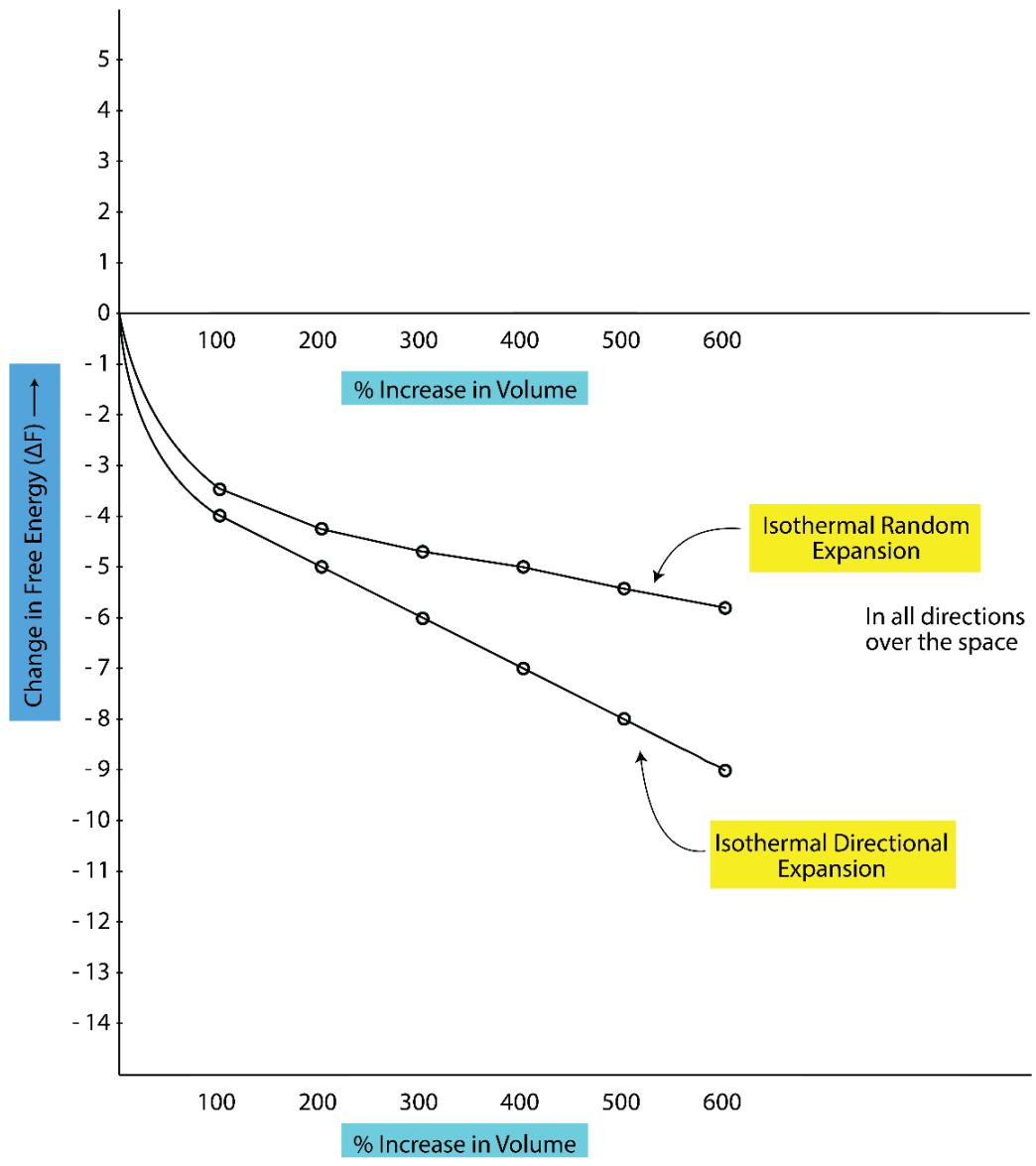 Figure 20: Change in 'Free Energy',
$\Delta F$ of 'Isothermal Random Expansion' vis-à-vis 'Isothermal Directional Expansion' From the data presented in Table 1.and the Figure20, one can understand, how much related is the free energy parameter to the topology of expansion of the system. For the directional expansion of the system, the free energy or the available 'thermodynamic work' is much higher than the homogeneous 'multi directional' expansion of the system.
## V. CONCLUSION
While dealing with the subject of 'Conventional Thermodynamics', a vast majority of the students and researchers find the subject un apprehensive since they do not find the proper answers of the many to many inquisitiveness persisting in their minds of the grey areas of this subject. As a result, especially the students, give off this subject at some stage or the other of their learning.
The 'time-space' version of thermodynamics, as is being presented here, in a very distinct and elogious manner opens up a new era in science and will be of immense help to the learners and especially the higher-level college/university students, the engineering students, for the true and in-depth understanding of the subject 'thermodynamics' and they will get proper answers of the grey areas of the subject and then will sit and move rather than giving off. Once this new concept are enrooted well in the mind of the readers, it will help them immensely to solve many practical real-life problems in physics/physical chemistry, biological science, geological science and many other streams too.
The basic text books of 'thermodynamics' need to be rewritten immediately for ending the time long carry over of misconceptions and that too for the advancement of science.
Dedication: I dedicate this research work to my beloved parents Late Mr. Krishna Pada Bhattacharya and Late Mrs. Aruna Bhattacharya and my very respectful brother in law Late Mr Hari Krishna Sannamat.
### ACKNOWLEDGEMENT
I do very sincerely acknowledge my wife Mrs. Chandana Bhattacharya, daughter Dr Sulagna Bhattacharya and the CEO, Mr Bijoy Sarkar, of M/S Austin Paints & Chemicals Private Limited, India for their high level encouragement and support to do the work.
I also acknowledge Mr Subhrojit Saikia and Mr Ai Myachha Chawloo of NIELIT, Jorhat Assam (India) for drawing the scientific diagrams of this work very sincerely.
Generating HTML Viewer...
Funding
No external funding was declared for this work.
Conflict of Interest
The authors declare no conflict of interest.
Ethical Approval
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
C. Bhattacharya. 2026. \u201cTheory of Space Quantization and the Quantum-Level Understanding of the True Logics of the Principles of Thermodynamics and Heat Engines\u201d. Global Journal of Science Frontier Research - A: Physics & Space Science GJSFR-A Volume 24 (GJSFR Volume 24 Issue A5): .
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
×
This Page is Under Development
We are currently updating this article page for a better experience.
Thank you for connecting with us. We will respond to you shortly.