Remodeling impacts of domain movements in protease are of interest in many fields such as medical treatments, food processing, and bio-electronic devices. However, they are yet to be precisely explained. In this study, the remodeling effects in ficin were investigated via a deep neural network, genetic programming, and computer simulations. The replacement (Y113F) in ficin using domain movements exhibited a critical effect on the peptide compatibilities. Specifically, modification of amino acid allows the remodeling of the domain movements, and types of compatible peptides should be modulated by the remodeling. Moreover, the decision tree revealed important factors in peptides and ficin.
## I. INTRODUCTION
Thermally produced movements of proteinous structure are perceived as the dominant factors involved in its biotic performance [1, 2, 3, 4], and their deliberate remodeling might lead to the discovery of an untapped resource. The majority of protein is organized via certain domain parts, and these domains are moving. Remodeling the domain movements would considerably influence molecular interactions between protein and ligand. Such interactions are of interest in many fields such as medical treatments, food processing, and bio-electronic devices [5, 6, 7, 8, 9], and their remodeling would provoke a breakthrough in these fields.
Hydrosoluble protein ficin separates certain peptides bound in sites near its active center, and its characteristics are beneficial to the improvement of transfusion safety, the development of novel antimicrobial therapeutics, and as a method for meat tenderization [10, 11, 12]. The structure of ficin is formed from two domains, and the binding areas of peptides are found in the two domains. Peptide binding is strongly affected by the domain movements, and therefore, remodeling the domain movements should affect the separation properties.
The two domains of ficin are joined by the $\beta$ strand, which acts as a hinge. Modification of amino acid in the $\beta$ strand would guide ficin to remodel the domain movements. It is desirable to be able to clearly explain the resulting effects of remodeling on peptide binding in more detail. Considering that ficin is a member of the protease family, reports on protease inhibitors play a large role in perceiving the effects [13, 14, 15, 16, 17]. However, remodeling effects of the domain movements in ficin to peptide bindings have not yet been clearly revealed.
The purpose of this research was to remodel the domain movements in ficin by modifying its local structure, and to clarify the effects of the remodeling to peptide bindings. Several possible combination patterns are possible for peptide, but the priority order of the search was unclear. The selection of combinations was conducted using deep neural networks (DNN) [18], and factors which enormously influence the peptide bindings were identified via genetic programming (GP) [19]. Characteristic features of various proteins were analyzed by molecular dynamics (MD) and docking simulations [20, 21, 13, 14, 22]. The moving structures of locally modified ficin were produced by MD simulation, while the binding states of peptides on the structures were assessed by docking simulations.
For the site near the center of the $\beta$ strand between the two domains of ficin, the amino acid was replaced by another one (Y113F). For the first trial, a similar amino acid (F) was selected for this replacement. In the previous study, the binding properties between the tetrapeptides and ficin were investigated by DNN, GP, MD and docking simulations [23]. Tetrapeptide structures have multiple structures, and they often have weights comparable to the protease inhibitor E64. In this study, the behavior of tetrapeptides on the replaced structure of ficin (ficin_Y113F) was researched using the dual artificial intelligences (AIs) and the simulations.
## II. METHODS
The selection speed of candidate peptides was better for DNN than for GP, and thus DNN was utilized to select the candidates for this pursuit. The factors that influence the peptide bindings need to be clarified, but they cannot be derived using DNN. Therefore, the factors were extracted using GP.
The potential candidates were selected through the use of a convolutional neural network (CNN) [24]. CNN is a type of DNN, and its structure was constructed using the deep learning library TensorFlow [25]. CNN is actively used in fields of image processing, and is capable of treating a large amount of partially reliable data. Its structure is composed of seven layers, namely, input layer, first convolutional layer, first pooling layer, second convolutional layer, second pooling layer, fully connected layer, and output layer. A tetrapeptide comprises a combination of four amino acids, and the amino acids contain multiple atoms. For each amino acid, each atom was numbered. The numbers were set in the input layer. For more stable binding states between tetrapeptide and sites near active center of ficin_Y113F, the distance between atoms in the peptides and its active center was calculated. The binding characteristics of peptides were classified by the respective numbers of atoms nearer than 4.0 Å to its active center. The classification rule was as follows: NA1≥3 atoms, 3 atoms>NA2≥2 atoms, 2 atoms>NA3≥1 atoms, or 1 atoms>NA4. The successive numbers from 1 to 4 were assigned to NAα ( $\alpha = 1-4$ ), and were set in the output layer. Further, the first ten tetrapeptides were derived from the DNN for ficin [23], and their binding properties to ficin_Y113F were investigated by the simulations. Thereafter, the first training data was created from the investigated results and training data of ficin [23]. Subsequently, the first DNN was constructed from the first training data, and consequently produced the second set of ten tetrapeptides. The properties of the second set of ten tetrapeptides were investigated in a similar manner, and second training data was made from the investigated results and the first training data. Consequently, the second DNN was constructed from the second training data, and subsequently produced the third ten tetrapeptides. Thus, this process was repeated, and various tetrapeptides were produced.
Highly Influencing factors to the peptide bindings were identified via GP. GP is an extended genetic algorithm (GA) technique, and it allows the exploration of the space of computer programs. The hyperparameters of GP were set with reference to a previous study [26], and evolutionary computing iteration was used to optimize the decision tree. The first time, entire areas of the decision tree were target of optimization. Thereafter, for the second and subsequent times, partial areas of the decision tree were randomly selected as optimization targets [27]. During the construction process of the decision tree, its size was expanded to satisfy the training data. Then, one of the successive numbers from 1 to 4 was set in the output of the decision tree, where the successive numbers corresponded to $\mathrm{NA}\alpha$ ( $\alpha = 1-4$ ). For a tetrapeptide, four amino acids were labeled as $\mathrm{ami}\beta$, $\beta = 1-4$ from the N-terminus to the C-terminus. Each amino acid ( $\mathrm{ami}\beta$ ) contained different types of atoms, and the number of each atom was represented by adding the atomic symbol at the end of $\mathrm{ami}\beta$. For instance, $\mathrm{ami4S}$ represented the number of sulfur atoms in amino acid of the C-terminus. The 20 attributes related to tetrapeptide were represented as $\mathrm{ami}\beta\mathrm{C}$, $\mathrm{ami}\beta\mathrm{N}$, $\mathrm{ami}\beta\mathrm{O}$, $\mathrm{ami}\beta\mathrm{S}$, and $\mathrm{ami}\beta\mathrm{AC}$ ( $\beta = 1-4$ ) [28]. 113 th amino acid of ficin was represented as $\mathrm{ami113fic}$, and the number of each atom in $\mathrm{ami113fic}$ was represented by adding the atomic symbol at the end of $\mathrm{ami113fic}$. The distance between domains of ficin was represented as domainDist, and it was assigned a numerical value of 0 for minimum distance, 1 for average distance, and 2 for maximum distance [23]. Further, 6 attributes related to ficin were represented as $\mathrm{ami113ficC}$, $\mathrm{ami113ficN}$, $\mathrm{ami113ficO}$, $\mathrm{ami113ficS}$, $\mathrm{ami113ficAC}$, and domainDist. 19 operations (Add, Sub, Mul, Div, Fmod, Log, Log10, Sin, Cos, If, Equal, NotEqual, GT, GE, And, Or, Not, Fmod2, and Sqrt) and the 12 constants (a constant integer number $\mathrm{CST}\gamma$ ( $\gamma = 0-9$ ) and a boolean value TRUE (1), FALSE (0)) were inherited from our previous studies [23, 27, 29, 28]. Furthermore, the construction factors of the decision tree were chosen from the 26 attributes, 19 operations, and 12 constants.
The structures of ficin_Y113F with movements were produced by MD simulation through the use of the packaged software AMBER 12.0 [30]. The initial coordinate data of ficin_Y113F was produced by homology modeling through the use of the software MODELLER [31, 32]. For homology modeling, structural prediction was performed from sequence homology with known structures. The produced structure of ficin_Y113F was solvated with 11,843 TIP3P water molecules [33] inside a rectangular box. For the solvated system, its temperature was gradually increased from 5 to $300\mathrm{K}$ over a period of 140 ps, and subsequently it was retained over a period of 100 ns until the precipitous structural changes vanished. Moreover, the ff03.r1 force field [34] was applied to manage MD simulation, and the hyperparameters were set by referencing the previous study [28]. In addition, the pressure and temperature were set using the Berendsen algorithm [35]. The long-distance electrostatic interactions were calculated using the particle mesh Ewald method [36]. The domain movements in ficin_Y113F were characterized as the distance variation between them for the period after the disappearance of precipitous structural changes. Thereafter, the average and snapshot structures were produced after the disappearance of the precipitous changes. The average structure was defined as $ficin\_Y113F\_ave$. Moreover, if the distance between the two domains of ficin_Y113F was at a maximum, the structure was defined as $ficin\_Y113F\_max$. Whereas, if the distance was at a minimum, the structure was defined as $ficin\_Y113F\_min$. The snapshot structures were made up of $ficin\_Y113F\_max$ and $ficin\_Y113F\_min$.
For ficin_Y113F_ave, ficin_Y113F_max and ficin_Y113F_min, binding affinities of the peptides were evaluated through the use of AutoDock Vina [37]. This simulation software was executed by using the iterated local search global optimizer algorithm [38, 39] and the Broyden-Fletcher-Goldfarb-Shanno method [40], with its scoring function derived by machine learning. These methods contributed significantly to approximately two orders of size speed-up compared to the molecular docking software AutoDock 4 [41, 42, 43]. Further, the coordinate data of the tetrapeptides was built by using the LEaP module in AMBER 12.0 [30]. The exploration space over the surface of ficin_Y113F was restricted to the regions near the active center. The explorations were executed 10 times for each tetrapeptide, and the most stable state was chosen from them. For the most stable state, atoms in the tetrapeptide were counted only if they were found near the active center of ficin_Y113F. However, if there were certain members in the most stable docking state, the counted numbers were averaged.
## III. RESULTS
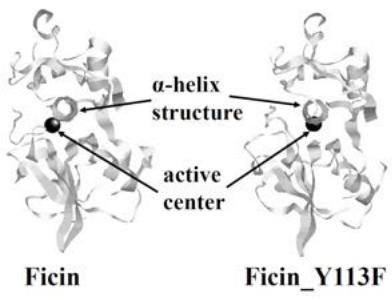
The temperature of the system was maintained at $300\mathrm{K}$ via MD simulations, and the precipitous structural changes of ficin_Y113F were surveyed at regular time intervals. For the structure of ficin_Y113F, the root-mean-square deviation (RMSD) between successive simulations was calculated to survey the precipitous changes(Figure S1). Consequently, the precipitous structural change was determined from the larger RMSD value, and they disappeared after 98,840 ps. The ficin_Y113F_ave was estimated in the range of 99,901 to 100,000 ps, and the snapshot structures (ficin_Y113F_max and ficin_Y113F_min) were derived from this range. Figure 1 shows the average structures of ficin and ficin_Y113F. The positional relation between the active center and the $\alpha$ -helix structure near the active center was changed by the replacement (Y113F). 10 tetrapeptides ((a) of Table S1) were selected as first candidates by using the data regarding ficin [23]. Thereafter, the candidates were docked to sites near active center of ficin_Y113F_ave, and the most stable styles were extracted.
Ficin is member of the family of cysteine protease, and the thiol (SH) group of its Cys25 can efficiently separate peptide. Consequently, the sulfur atom of Cys25 (S-Cys25) was considered as the active center. To discover the talented peptides for ficin_Y113F, the number of the atoms nearer than 4.0 Å to S-Cys25 in the most-stable docking state were estimated. In case of certain most-stable states, the estimated numbers were averaged. These results were represented by $\mathrm{NA}\alpha$ ( $\alpha = 1 - 4$ ), and are listed in Table S2. Within the first ten peptides, one member (Asn-Ser-Lys-Gln) was compatible with the sites near S-Cys25 of ficin_Y113F. Moreover, machine learning of the CNN was executed with the addition of these results, and the next ten candidates ((b) of Table S1) were guided. The characteristics of the ten peptides were estimated by docking simulations (Table S2), and one member (Arg-Ala-Val-Val) was compatible with the sites. This process was repeated eight times, and thereby, the characteristics of 80 peptides were obtained (Table S2), within which 17 members were compatible. The compatible 17 peptides were deposited in ave of Table 1. Simultaneously, 38 members were identified as especially incompatible peptides (Table S3). The docking simulations of the compatible 17 peptides (ave of Table 1) to the sites near S-Cys25 of two snapshot structures (ficin_Y113F_max and ficin_Y113F_min) were executed, and the results are presented in Table S4. Within the 17 peptides, nine members (max of Table 1) were compatible to the sites near S-Cys25 of ficin_Y113F_max and six members (min of Table 1) were compatible to those of ficin_Y113F_min. Furthermore, the five members (both of Table 1) were compatible to them of both ficin_Y113F_max and ficin_Y113F_min.
To determine the important factors related to the peptide bindings, a decision tree was constructed from the results of 17 tetrapeptides to the three states of ficin_Y113F (ficin_Y113F_min, ficin_Y113F_ave, and ficin_Y113F_max)(Table 1) and those of 4 tetrapeptides to the three states of non-modified ficin (ficin_min, ficin_ave, and ficin_max) [23]. The decision tree comprised 45 nodes, and its layout is indicated in Table 2 and Table 3. 11 attributes were included in the decision tree, and these were considered as the important factors. They were one ami1N, one ami1O, one ami1S, two ami2C, one ami3C, two ami3N, one ami113ficO, and two domainDist.
The acronyms were explained in Table S5.
## IV. DISCUSSION
The compatibilities of the 100 tetrapeptides to the sites near S-Cys25 of $ficin\_Y113F\_ave$ were revealed via a DNN, and MD and docking simulations. The 17 compatible peptides were found, and about 47 percent of them were not compatible for $ficin\_Y113F\_max$, and about 65 percent of them were not compatible for $ficin\_Y113F\_min$ (Table 1). The compatibilities of the tetrapeptides to the sites near S-Cys25 of $ficin\_Y113F$ were grossly influenced by their domain movements. The five peptides were found to be compatible for the three states of $ficin\_Y113F$ (Table 1). The five peptides were not influenced by the domain movements of $ficin\_Y113F$, and their structures would be an informative guide to drug development.
For average structure of non-modified ficin (ficin_ave), only four peptides were compatible to the sites near S-Cys25 [23]. The replacement (Y113F) in ficin spread the compatible peptides within those of ficin_ave. Within the four peptides, only one peptide was compatible for the structure of non-modified ficin when the distance between their domains was at a maximum (ficin_max) [23]. In contrast, the replacement (Y113F) spread the compatible peptides within those of ficin_max as well. Further, all the four peptides were not compatible for the structure of non-modified ficin when the distance between their domains was at a minimum (ficin_min) [23]. The replacement (Y113F) spread the compatible peptides within those of ficin_min as well as above two structures and had significant influence on the compatibilities between the peptides and ficin with domain movements. Moreover, modification of amino acid in the hinge structure of protease can enable us to remodel the domain movements, while the types of compatible peptides should be regulated by the remodeling. The collaboration of a DNN, and MD and docking simulations can be utilized as a tool in clarification of induced effects by modification of domain movements in protease.
The decision tree was constructed to satisfy the results for the three structures of ficin_Y113F (ficin_Y113 F_min, ficin_Y113Eave, and ficin_Y113Emax) (Table 1) and also for the three structures of non-modified ficin (ficin_min, ficin_ave, and ficin_max) [23]. The tree was composed of 45 nodes, and the breakdown of its 45 nodes was 11 attributes, 25 operations, and 9 constants (Table 2 and Table 3). The 11 attributes included one ami1N, one ami1O, one ami1S, two ami2C, one ami3C, two ami3N, one ami13ficO, and two domainDist. Carbon atoms in second amino acid from the N-terminus of the peptide, nitrogen atoms in third amino acid from the N-terminus of it, and domain movements of the protein are the important factors governing the peptide compatibility. The three atoms (nitrogen, oxygen, and sulfur) in the N-terminus amino acid of the peptide, carbon atom in third amino acid from the N-terminus of it, and oxygen atom in 113 th amino acid in the protein must also be focused on. The 25 operations included four Add, three Sub, seven Mul, three Div, two Fmod, one Fmod2, three Sqrt, and two Sin. Many of the operations were the four basic arithmetic operations, and the peptide compatibility can be derived from comparatively elementary manner. The number of Sqrt was also relatively large, and numerical value could be be moderately changed. Furthermore, the logical operations were not included, and the derivation of the compatibility could not be expressed in binary form.
## V. CONCLUSIONS
The remodeling effects of the domain movements in ficin were analyzed by using DNN, GP, MD, and docking simulations. The replacement (Y113F) in ficin had critical effect on the compatibilities between the peptides and ficin with domain movements. Further, modification of specific amino acid in protease can enable remodeling of the domain movements, and types of compatible peptides must be modulated by the remodeling. Moreover, the decision tree revealed that particular atom in particular amino acid in peptide and domain movements of the protease would be of central importance for the peptide compatibility. Furthermore, the tree indicated the contribution of oxygen atom in 113 th amino acid in the protease to the compatibility. Thus, the analytical approach consisting of DNN, GP, MD and docking simulations can offer valuable lessons to understand the remodeling effects of the domain movements for some structures of protease, and the other structures should be analyzed for further research.
### ACKNOWLEDGMENTS
This study was supported by the National Institute of Technology (KOSEN).
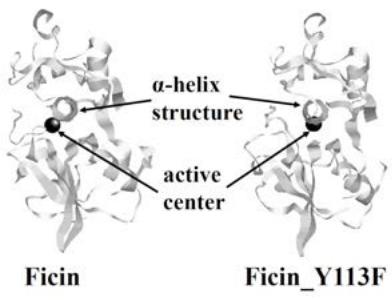 Figure 1: The average structures of ficin and ficin_Y113F.
Table 1: The compatible peptides.
<table><tr><td>ave</td><td>max</td><td>min</td><td>both</td></tr><tr><td>Arg-Ala-Val-Val</td><td></td><td></td><td></td></tr><tr><td>Arg-Asp-Lys-Arg</td><td>○</td><td>○</td><td>○</td></tr><tr><td>Arg-Gly-Arg-Gln</td><td>○</td><td></td><td></td></tr><tr><td>Arg-Gly-Asn-Ile</td><td>○</td><td>○</td><td>○</td></tr><tr><td>Arg-Gly-Asn-Leu</td><td>○</td><td>○</td><td>○</td></tr><tr><td>Arg-Gly-Asn-Lys</td><td>○</td><td>○</td><td>○</td></tr><tr><td>Arg-Gly-Asn-Val</td><td>○</td><td>○</td><td>○</td></tr><tr><td>Arg-Gly-Gln-Lys</td><td></td><td></td><td></td></tr><tr><td>Arg-Gly-Pro-Ile</td><td>○</td><td></td><td></td></tr><tr><td>Arg-Gly-Pro-Leu</td><td></td><td></td><td></td></tr><tr><td>Arg-Gly-Ser-Leu</td><td>○</td><td></td><td></td></tr><tr><td>Arg-Gly-Thr-Leu</td><td></td><td></td><td></td></tr><tr><td>Arg-Gly-Val-Leu</td><td>○</td><td></td><td></td></tr><tr><td>Arg-Ser-Leu-Lys</td><td></td><td></td><td></td></tr><tr><td>Asn-Ser-Lys-Gln</td><td></td><td>○</td><td></td></tr><tr><td>Lys-Gly-Leu-Gln</td><td></td><td></td><td></td></tr><tr><td>Lys-Ser-Ile-Arg</td><td></td><td></td><td></td></tr></table>
Table 2: Layout of first part of decision tree (α:Input, β:Output, γ:Node).
<table><tr><td>γ0</td><td>Fmod2[α0 = β of γ1][β = Solution]</td></tr><tr><td>γ1</td><td>Fmod[α0 = β of γ2][α1 = β of γ33][β = α0 of γ0]</td></tr><tr><td>γ2</td><td>Mul[α0 = β of γ3][α1 = β of γ32][β = α0 of γ1]</td></tr><tr><td>γ3</td><td>Fmod[α0 = β of γ4][α1 = β of γ27][β = α0 of γ2]</td></tr><tr><td>γ4</td><td>Mul[α0 = β of γ5][α1 = β of γ6][β = α0 of γ3]</td></tr><tr><td>γ5</td><td>ami3N[β = α0 of γ4]</td></tr><tr><td>γ6</td><td>Sub[α0 = β of γ7][α1 = β of γ8][β = α1 of γ4]</td></tr><tr><td>γ7</td><td>domainDist[β = α0 of γ6]</td></tr><tr><td>γ8</td><td>Mul[α0 = β of γ9][α1 = β of γ20][β = α1 of γ6]</td></tr><tr><td>γ9</td><td>Add[α0 = β of γ10][α1 = β of γ18][β = α0 of γ8]</td></tr><tr><td>γ10</td><td>Mul[α0 = β of γ11][α1 = β of γ17][β = α0 of γ9]</td></tr><tr><td>γ11</td><td>Mul[α0 = β of γ12][α1 = β of γ16][β = α0 of γ10]</td></tr><tr><td>γ12</td><td>Div[α0 = β of γ13][α1 = β of γ15][β = α0 of γ11]</td></tr><tr><td>γ13</td><td>Sin[α0 = β of γ14][β = α0 of γ12]</td></tr><tr><td>γ14</td><td>Cst2[β = α0 of γ13]</td></tr><tr><td>γ15</td><td>domainDist[β = α1 of γ12]</td></tr><tr><td>γ16</td><td>ami2C[β = α1 of γ11]</td></tr><tr><td>γ17</td><td>Cst6[β = α1 of γ10]</td></tr><tr><td>γ18</td><td>Sqrt[α0 = β of γ19][β = α1 of γ9]</td></tr><tr><td>γ19</td><td>Cst3[β = α0 of γ18]</td></tr><tr><td>γ20</td><td>Add[α0 = β of γ21][α1 = β of γ25][β = α1 of γ8]</td></tr><tr><td>γ21</td><td>Mul[α0 = β of γ22][α1 = β of γ24][β = α0 of γ20]</td></tr><tr><td>γ22</td><td>Sqrt[α0 = β of γ23][β = α0 of γ21]</td></tr></table>
Table 3: Layout of second part of decision tree (α:Input, β:Output, γ:Node).
<table><tr><td>γ23</td><td>Cst4[β = α0 of γ22]</td></tr><tr><td>γ24</td><td>Cst7[β = α1 of γ21]</td></tr><tr><td>γ25</td><td>Sqrt[α0 = β of γ26][β = α1 of γ20]</td></tr><tr><td>γ26</td><td>Cst5[β = α0 of γ25]</td></tr><tr><td>γ27</td><td>Sub[α0 = β of γ28][α1 = β of γ31][β = α1 of γ3]</td></tr><tr><td>γ28</td><td>Mul[α0 = β of γ29][α1 = β of γ30][β = α0 of γ27]</td></tr><tr><td>γ29</td><td>ami1O[β = α0 of γ28]</td></tr><tr><td>γ30</td><td>ami3C[β = α1 of γ28]</td></tr><tr><td>γ31</td><td>ami1S[β = α1 of γ27]</td></tr><tr><td>γ32</td><td>Cst2[β = α1 of γ2]</td></tr><tr><td>γ33</td><td>Add[α0 = β of γ34][α1 = β of γ35][β = α1 of γ1]</td></tr><tr><td>γ34</td><td>Cst6[β = α0 of γ33]</td></tr><tr><td>γ35</td><td>Div[α0 = β of γ36][α1 = β of γ37][β = α1 of γ33]</td></tr><tr><td>γ36</td><td>ami2C[β = α0 of γ35]</td></tr><tr><td>γ37</td><td>Div[α0 = β of γ38][α1 = β of γ43][β = α1 of γ35]</td></tr><tr><td>γ38</td><td>Add[α0 = β of γ39][α1 = β of γ40][β = α0 of γ37]</td></tr><tr><td>γ39</td><td>ami3N[β = α0 of γ38]</td></tr><tr><td>γ40</td><td>Sub[α0 = β of γ41][α1 = β of γ42][β = α1 of γ38]</td></tr><tr><td>γ41</td><td>ami113ficO[β = α0 of γ40]</td></tr><tr><td>γ42</td><td>Cst1[β = α1 of γ40]</td></tr><tr><td>γ43</td><td>Sin[α0 = β of γ44][β = α1 of γ37]</td></tr><tr><td>γ44</td><td>ami1N[β = α0 of γ43]</td></tr></table>
#### Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version at
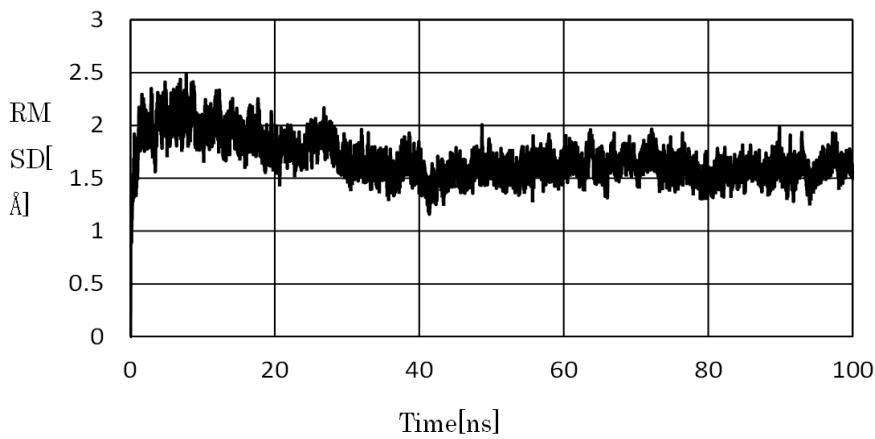 Figure S1.: RMSD between the successive simulations.
Table S1: The selected tetrapeptides.
<table><tr><td>(a)</td><td>(b)</td></tr><tr><td>Ser-Ser-Ile-Arg</td><td>Met-Gly-Lys-Gln</td></tr><tr><td>Arg-Cys-Lys-Arg</td><td>Lys-Cys-Arg-Gln</td></tr><tr><td>His-Cys-Arg-Ala</td><td>Gln-Ser-Arg-Asn</td></tr><tr><td>Arg-Asn-Arg-Asn</td><td>Arg-Ser-Lys-Lys</td></tr><tr><td>Thr-Asn-Arg-Arg</td><td>Gln-Ala-Arg-Asn</td></tr><tr><td>His-Gly-Leu-Gly</td><td>Arg-Ala-Val-Val</td></tr><tr><td>Arg-Gln-Arg-Arg</td><td>Lys-Gly-Lys-Val</td></tr><tr><td>Asn-Ser-Lys-Gln</td><td>His-Gly-Arg-Asn</td></tr><tr><td>Leu-Gly-Arg-Asn</td><td>Arg-Asp-Arg-Pro</td></tr><tr><td>Met-Asp-Ile-Arg</td><td>Lys-Ala-Lys-Lys</td></tr></table>
Table S2: Affinity of the 100 tetrapeptides in the active region of the average structure of locally modified ficin (Y113F).
<table><tr><td>Ser-Ser-Ile-Arg</td><td>NA3</td><td>Lys-Gly-Ile-Gln</td><td>NA4</td><td>Arg-Gly-Lys-Leu</td><td>NA2</td><td>Arg-Gly-Lys-Asn</td><td>NA4</td></tr><tr><td>Arg-Cys-Lys-Arg</td><td>NA2</td><td>Gln-Gly-Ile-Gln</td><td>NA4</td><td>Gln-Gly-Lys-Lys</td><td>NA4</td><td>Arg-Gly-Arg-Thr</td><td>NA4</td></tr><tr><td>His-Cys-Arg-Ala</td><td>NA3</td><td>Gln-Gly-Leu-Gln</td><td>NA4</td><td>Arg-Gly-Gln-Leu</td><td>NA2</td><td>Arg-Gly-Arg-Asn</td><td>NA2</td></tr><tr><td>Arg-Asn-Arg-Asn</td><td>NA2</td><td>Lys-Gly-Val-Gln</td><td>NA4</td><td>Arg-Gly-Asn-Leu</td><td>NA1</td><td>Val-Trp-Cys-Gly</td><td>NA4</td></tr><tr><td>Thr-Asn-Arg-Arg</td><td>NA3</td><td>Arg-Gly-Val-Leu</td><td>NA1</td><td>Arg-Gly-Ser-Lys</td><td>NA4</td><td>Met-Arg-Ile-Phe</td><td>NA4</td></tr><tr><td>His-Gly-Leu-Gly</td><td>NA4</td><td>Lys-Gly-Ile-Leu</td><td>NA4</td><td>Arg-Gly-Pro-Lys</td><td>NA3</td><td>Met-Gly-Glu-Arg</td><td>NA3</td></tr><tr><td>Arg-Gln-Arg-Arg</td><td>NA3</td><td>Arg-Gly-Lys-Met</td><td>NA3</td><td>Met-Gly-Val-Lys</td><td>NA4</td><td>Arg-Gly-Glu-Leu</td><td>NA4</td></tr><tr><td>Asn-Ser-Lys-Gln</td><td>NA1</td><td>Lys-Gly-Val-Leu</td><td>NA3</td><td>Arg-Gly-Ala-Leu</td><td>NA3</td><td>Lys-Gly-Gln-Arg</td><td>NA4</td></tr><tr><td>Leu-Gly-Arg-Asn</td><td>NA4</td><td>Arg-Gly-Val-Gln</td><td>NA2</td><td>Arg-Gly-Lys-Gln</td><td>NA3</td><td>Arg-Gly-Thr-Ile</td><td>NA3</td></tr><tr><td>Met-Asp-Ile-Arg</td><td>NA3</td><td>Arg-Gly-Arg-Gln</td><td>NA1</td><td>Arg-Gly-Gln-Gln</td><td>NA4</td><td>Ile-Gly-Asp-Arg</td><td>NA4</td></tr><tr><td>Met-Gly-Lys-Gln</td><td>NA3</td><td>Arg-Gly-Pro-Gln</td><td>NA4</td><td>Arg-Gly-Gln-Ile</td><td>NA4</td><td>Arg-Gly-Asp-Leu</td><td>NA4</td></tr><tr><td>Lys-Cys-Arg-Gln</td><td>NA4</td><td>Arg-Gly-Arg-Lys</td><td>NA3</td><td>Arg-Ser-Lys-Ile</td><td>NA3</td><td>Ile-Gly-Glu-Arg</td><td>NA4</td></tr><tr><td>Gln-Ser-Arg-Asn</td><td>NA4</td><td>Lys-Gly-Pro-Ile</td><td>NA2</td><td>Arg-Ser-Lys-Leu</td><td>NA4</td><td>Arg-Gly-Gln-Lys</td><td>NA1</td></tr><tr><td>Arg-Ser-Lys-Lys</td><td>NA2</td><td>Arg-Gly-Leu-Lys</td><td>NA4</td><td>Arg-Ala-Gln-Leu</td><td>NA2</td><td>Arg-Gly-Thr-Leu</td><td>NA1</td></tr><tr><td>Gln-Ala-Arg-Asn</td><td>NA3</td><td>Arg-Ala-Gln-Lys</td><td>NA4</td><td>Arg-Gly-Asn-Ile</td><td>NA1</td><td>Leu-Gly-Glu-Arg</td><td>NA3</td></tr><tr><td>Arg-Ala-Val-Val</td><td>NA1</td><td>Arg-Gly-Leu-Val</td><td>NA4</td><td>Gln-Asp-Lys-Arg</td><td>NA3</td><td>Arg-Gly-Ser-Ile</td><td>NA2</td></tr><tr><td>Lys-Gly-Lys-Val</td><td>NA4</td><td>Arg-Cys-Leu-Leu</td><td>NA4</td><td>Lys-Ser-Leu-Arg</td><td>NA3</td><td>Arg-Gly-Ser-Leu</td><td>NA1</td></tr><tr><td>His-Gly-Arg-Asn</td><td>NA4</td><td>Arg-Ser-Leu-Lys</td><td>NA1</td><td>Arg-Asp-Lys-Arg</td><td>NA1</td><td>Arg-Gly-Ile-Lys</td><td>NA3</td></tr><tr><td>Arg-Asp-Arg-Pro</td><td>NA4</td><td>Arg-Ser-Ile-Lys</td><td>NA4</td><td>Gln-Asp-Ile-Arg</td><td>NA4</td><td>Arg-Gly-Val-Arg</td><td>NA4</td></tr><tr><td>Lys-Ala-Lys-Lys</td><td>NA4</td><td>Arg-Gly-Leu-Ile</td><td>NA2</td><td>Lys-Ser-Ile-Arg</td><td>NA1</td><td>Arg-Ser-Gln-Arg</td><td>NA4</td></tr><tr><td>Arg-Gly-Val-Ile</td><td>NA3</td><td>Arg-Gly-Asn-Lys</td><td>NA1</td><td>Arg-Cys-Lys-Lys</td><td>NA2</td><td>Arg-Ser-Val-Arg</td><td>NA2</td></tr><tr><td>Arg-Gly-Pro-Leu</td><td>NA1</td><td>Arg-Gly-Lys-Pro</td><td>NA3</td><td>Arg-Gly-Thr-Lys</td><td>NA2</td><td>Arg-Ser-Ile-Arg</td><td>NA3</td></tr><tr><td>Lys-Gly-Pro-Gln</td><td>NA3</td><td>Arg-Ala-Leu-Lys</td><td>NA3</td><td>Arg-Gly-Asn-Val</td><td>NA1</td><td>Arg-Ser-Leu-Arg</td><td>NA3</td></tr><tr><td>Lys-Gly-Leu-Gln</td><td>NA1</td><td>Arg-Gly-Lys-Lys</td><td>NA3</td><td>Arg-Ala-Lys-Asn</td><td>NA4</td><td>Arg-Ser-Pro-Arg</td><td>NA2</td></tr><tr><td>Arg-Gly-Pro-Ile</td><td>NA1</td><td>Arg-Gly-Ile-Met</td><td>NA3</td><td>Arg-Cys-Arg-Asn</td><td>NA3</td><td>Arg-Gly-Lys-Arg</td><td>NA3</td></tr></table>
Table S3: The 38 especially incompatible tetrapeptides.
<table><tr><td>His-Gly-Leu-Gly</td><td>Gln-Gly-Lys-Lys</td></tr><tr><td>Leu-Gly-Arg-Asn</td><td>Arg-Gly-Ser-Lys</td></tr><tr><td>Lys-Cys-Arg-Gln</td><td>Met-Gly-Val-Lys</td></tr><tr><td>Gln-Ser-Arg-Asn</td><td>Arg-Gly-Gln-Gln</td></tr><tr><td>Lys-Gly-Lys-Val</td><td>Arg-Gly-Gln-Ile</td></tr><tr><td>His-Gly-Arg-Asn</td><td>Arg-Ser-Lys-Leu</td></tr><tr><td>Arg-Asp-Arg-Pro</td><td>Gln-Asp-Ile-Arg</td></tr><tr><td>Lys-Ala-Lys-Lys</td><td>Arg-Ala-Lys-Asn</td></tr><tr><td>Lys-Gly-Ile-Gln</td><td>Arg-Gly-Lys-Asn</td></tr><tr><td>Gln-Gly-Ile-Gln</td><td>Arg-Gly-Arg-Thr</td></tr><tr><td>Gln-Gly-Leu-Gln</td><td>Val-Trp-Cys-Gly</td></tr><tr><td>Lys-Gly-Val-Gln</td><td>Met-Arg-Ile-Phe</td></tr><tr><td>Lys-Gly-Ile-Leu</td><td>Arg-Gly-Glu-Leu</td></tr><tr><td>Arg-Gly-Pro-Gln</td><td>Lys-Gly-Gln-Arg</td></tr><tr><td>Arg-Gly-Leu-Lys</td><td>Ile-Gly-Asp-Arg</td></tr><tr><td>Arg-Ala-Gln-Lys</td><td>Arg-Gly-Asp-Leu</td></tr><tr><td>Arg-Gly-Leu-Val</td><td>Ile-Gly-Glu-Arg</td></tr><tr><td>Arg-Cys-Leu-Leu</td><td>Arg-Gly-Val-Arg</td></tr><tr><td>Arg-Ser-Ile-Lys</td><td>Arg-Ser-Gln-Arg</td></tr></table>
Table S4: Affinity of the 17 tetrapeptides in the active region of the snapshot structures of locally modified ficin (Y113F).
<table><tr><td></td><td>max</td><td>min</td></tr><tr><td>Arg-Ala-Val-Val</td><td>NA3</td><td>NA3</td></tr><tr><td>Arg-Asp-Lys-Arg</td><td>NA1</td><td>NA1</td></tr><tr><td>Arg-Gly-Arg-Gln</td><td>NA1</td><td>NA3</td></tr><tr><td>Arg-Gly-Asn-Ile</td><td>NA1</td><td>NA1</td></tr><tr><td>Arg-Gly-Asn-Leu</td><td>NA1</td><td>NA1</td></tr><tr><td>Arg-Gly-Asn-Lys</td><td>NA1</td><td>NA1</td></tr><tr><td>Arg-Gly-Asn-Val</td><td>NA1</td><td>NA1</td></tr><tr><td>Arg-Gly-Gln-Lys</td><td>NA2</td><td>NA3</td></tr><tr><td>Arg-Gly-Pro-Ile</td><td>NA1</td><td>NA3</td></tr><tr><td>Arg-Gly-Pro-Leu</td><td>NA3</td><td>NA3</td></tr><tr><td>Arg-Gly-Ser-Leu</td><td>NA1</td><td>NA3</td></tr><tr><td>Arg-Gly-Thr-Leu</td><td>NA3</td><td>NA4</td></tr><tr><td>Arg-Gly-Val-Leu</td><td>NA1</td><td>NA3</td></tr><tr><td>Arg-Ser-Leu-Lys</td><td>NA2</td><td>NA3</td></tr><tr><td>Asn-Ser-Lys-Gln</td><td>NA3</td><td>NA1</td></tr><tr><td>Lys-Gly-Leu-Gln</td><td>NA3</td><td>NA3</td></tr><tr><td>Lys-Ser-Ile-Arg</td><td>NA2</td><td>NA3</td></tr></table>
Table S5: Explanation of the acronyms. (α: Input β: Output).
<table><tr><td>Add[α0][α1][β]</td><td>β = arithmetic addition of α0 and α1</td></tr><tr><td>Sub[α0][α1][β]</td><td>β = subtraction of α1 from α0</td></tr><tr><td>Mul[α0][α1][β]</td><td>β = arithmetic multiplication of α0 and α1</td></tr><tr><td>Div[α0][α1][β]</td><td>β = division of α0 by α1</td></tr><tr><td>Fmod[α0][α1][β]</td><td>β = remainder after division of α0 by α1</td></tr><tr><td>Log[α0][β]</td><td>β = natural logarithm of α0</td></tr><tr><td>Log10[α0][β]</td><td>β = common logarithm of α0</td></tr><tr><td>Sin[α0][α1][α2][β]</td><td>β = arithmetic multiplication of α0 and X, where X is sine of division of α1 by α2</td></tr><tr><td>Cos[α0][α1][α2][β]</td><td>β = arithmetic multiplication of α0 and X, where X is cosine of division of α1 by α2</td></tr><tr><td>If[α0][α1][α2][β]</td><td>β = if α0 = true then α1 else α2</td></tr><tr><td>Equal[α0][α1][β]</td><td>β = if α0 = α1 then true else false</td></tr><tr><td>NotEqual[α0][α1][β]</td><td>β = if α0 = α1 then false else true</td></tr><tr><td>GT[α0][α1][β]</td><td>β = if α0 > α1 then true else false</td></tr><tr><td>GE[α0][α1][β]</td><td>β = if α0 ≥ α1 then true else false</td></tr><tr><td>And[α0][α1][β]</td><td>β = logical multiplication of α0 and α1</td></tr><tr><td>Or[α0][α1][β]</td><td>β = logical addition of α0 and α1</td></tr><tr><td>Not[α0][β]</td><td>β = logical negation of α0</td></tr><tr><td>Fmod2[α0][β]</td><td>β = remainder after division of α0 by 4</td></tr><tr><td>Sqrt[α0][β]</td><td>β = square root of α0</td></tr><tr><td>ami γ</td><td>4 amino acids in a tetrapeptide, γ = 1-4 from the N-terminus to the C-terminus</td></tr><tr><td>ami γC</td><td>the number of carbon atoms in ami γ</td></tr><tr><td>ami γN</td><td>the number of nitrogen atoms in ami γ</td></tr><tr><td>ami γO</td><td>the number of oxygen atoms in ami γ</td></tr><tr><td>ami γS</td><td>the number of sulfur atoms in ami γ</td></tr><tr><td>ami γAC</td><td>the number of aromatic carbon atoms in ami γ</td></tr><tr><td>ami113ficC</td><td>the number of carbon atoms in 113~th amino acid of ficin</td></tr><tr><td>ami113ficN</td><td>the number of nitrogen atoms in 113~th amino acid of ficin</td></tr><tr><td>ami113ficO</td><td>the number of oxygen atoms in 113~th amino acid of ficin</td></tr><tr><td>ami113ficS</td><td>the number of sulfur atoms in 113~th amino acid of ficin</td></tr><tr><td>ami113ficAC</td><td>the number of aromatic carbon atoms in 113~th amino acid of ficin</td></tr><tr><td>domainDist</td><td>the distance between domains of ficin (0:minimum, 1:average, 2:maximum)</td></tr><tr><td>CST δ</td><td>a constant integer number δ (δ = 0-9)</td></tr><tr><td>TRUE</td><td>a boolean value 1</td></tr><tr><td>FALSE</td><td>a boolean value 0</td></tr><tr><td>ficin_Y113F_ave</td><td>the average structure of ficin_Y113F</td></tr><tr><td>ficin_Y113F_max</td><td>the snapshot structure of ficin_Y113F when distance between its domains is at a maximum</td></tr><tr><td>ficin_Y113F-mini</td><td>the snapshot structure of ficin_Y113F when distance between its domains is at a minimum</td></tr></table>
N Lys-Ala-Lys-Lys (2019). Orphan designation: Tyr-Met-Phe-Pro-Asn-Ala-Pro-Tyr-Leu, Ser-Gly-Gln-Ala-Tyr-Met-Phe-Pro-Asn-Ala-Pro-Tyr-Leu-Pro-Ser-Cys-Leu-Glu-Ser, Arg-Ser-Asp-Glu-Leu-Val-Arg-His-His-Asn-Met-His-Gln-Arg-Asn-Met-Thr-Lys-Leu and Pro-Gly-Cys-Asn-Lys-Arg-Tyr-Phe-Lys-Leu-Ser-His-Leu-Gln-Met-His-Ser-Arg-Lys-His-Thr-Gly, Treatment of malignant mesothelioma.
D Li,D Meng,R Bruschweiler (2009). Unknown Title.
Andrea Amadei,Antonius Linssen,Herman Berendsen (1993). Essential dynamics of proteins.
J Mccammon,Bruce Gelin,Martin Karplus,Peter Wolynes (1976). The hinge-bending mode in lysozyme.
Juan Yguerabide,Henry Epstein,Lubert Stryer (1970). Segmental flexibility in an antibody molecule.
Huihua Sun,Hongfang Zhang,Ee Ang,Huimin Zhao (2018). Biocatalysis for the synthesis of pharmaceuticals and pharmaceutical intermediates.
A Bezborodov,N Zagustina (2016). Enzymatic biocatalysis in chemical synthesis of pharmaceuticals (Review).
Sindhu Raveendran,Binod Parameswaran,Sabeela Ummalyma,Amith Abraham,Anil Mathew,Aravind Madhavan,Sharrel Rebello,Ashok Pandey (2018). Applications of Microbial Enzymes in Food Industry.
Takashi Tanii,Tomomi Goto,Tomoyuki Iida,Meishoku Koh-Masahara,Iwao Ohdomari (2001). Fabrication of Adenosine Triphosphate-Molecule Recognition Chip by Means of Bioluminous Enzyme Luciferase.
Hiroyasu Itoh,Akira Takahashi,Kengo Adachi,Hiroyuki Noji,Ryohei Yasuda,Masasuke Yoshida,Kazuhiko Kinosita (2004). Mechanically driven ATP synthesis by F1-ATPase.
Ben Hill,Courtney Hanna,Jill Adamski,Huy Pham,Marisa Marques,Lance Williams (2016). Ficin-Treated Red Cells Help Identify Clinically Significant Alloantibodies Masked as Reactions of Undetermined Specificity in Gel Microtubes.
G Walsh (2002). Unknown Title.
Shou Matsuyama,Ay Aydan,Hirotaka Ode,Masayuki Hata,Wataru Sugiura,Tyuji Hoshino (2010). Structural and Energetic Analysis on the Complexes of Clinically Isolated Subtype C HIV-1 Proteases and Approved Inhibitors by Molecular Dynamics Simulation.
Hirotaka Ode,Saburo Neya,Masayuki Hata,Wataru Sugiura,Tyuji Hoshino (2006). Computational Simulations of HIV-1 ProteasesMulti-drug Resistance Due to Nonactive Site Mutation L90M.
P Drabik,E Politowska,C Czaplewski,F Kasprzykowski,L Lankiewicz,J Ciarkowski (2000). Theoretical studies of binding modes of two covalent inhibitors of cysteine proteases..
X Hu,S Balaz,W Shelver (2004). Unknown Title.
Fredrik Österberg,Garrett Morris,Michel Sanner,Arthur Olson,David Goodsell (2002). Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock.
David Silver,Aja Huang,Chris Maddison,Arthur Guez,Laurent Sifre,George Van Den Driessche,Julian Schrittwieser,Ioannis Antonoglou,Veda Panneershelvam,Marc Lanctot,Sander Dieleman,Dominik Grewe,John Nham,Nal Kalchbrenner,Ilya Sutskever,Timothy Lillicrap,Madeleine Leach,Koray Kavukcuoglu,Thore Graepel,Demis Hassabis (2016). Mastering the game of Go with deep neural networks and tree search.
Johnr. Koza (1992). Genetic programming as a means for programming computers by natural selection.
Hiroshi Yanagita,Norio Yamamoto,Hideyoshi Fuji,Xinli Liu,Masakazu Ogata,Mizuho Yokota,Hiroshi Takaku,Hideki Hasegawa,Takato Odagiri,Masato Tashiro,Tyuji Hoshino (2012). Mechanism of Drug Resistance of Hemagglutinin of Influenza Virus and Potent Scaffolds Inhibiting Its Function.
Hitomi Yuki,Teruki Honma,Masayuki Hata,Tyuji Hoshino (2012). Prediction of sites of metabolism in a substrate molecule, instanced by carbamazepine oxidation by CYP3A4.
Katsuhiko Nishiyama (2016). Decision tree for the binding of dipeptides to the thermally fluctuating surface of cathepsin K.
Marc Martí-Renom,Ashley Stuart,András Fiser,Roberto Sánchez,Francisco Melo,Andrej Šali (2000). Comparative Protein Structure Modeling of Genes and Genomes.
W Jorgensen,J Chandrasekhar,J Madura (1983). Unknown Title.
Yong Duan,Chun Wu,Shibasish Chowdhury,Mathew Lee,Guoming Xiong,Wei Zhang,Rong Yang,Piotr Cieplak,Ray Luo,Taisung Lee,James Caldwell,Junmei Wang,Peter Kollman (2003). A point‐charge force field for molecular mechanics simulations of proteins based on condensed‐phase quantum mechanical calculations.
H Berendsen,J Postma,W Gunsteren,A Dinola,J Haak (1984). Unknown Title.
Tom Darden,Darrin York,Lee Pedersen (1993). Particle mesh Ewald: An <i>N</i>⋅log(<i>N</i>) method for Ewald sums in large systems.
Oleg Trott,Arthur Olson (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.
John Baxter (1981). Local Optima Avoidance in Depot Location.
C Blum,A Roli,M Sampels (2008). Hybrid Metaheuristics: An Emerging Approach to Optimization.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Katsuhiko Nishiyama. 2026. \u201cInspection of Remodeling Impacts of Domain Movements in Hydrosoluble Protein Using Dual Artificial Intelligence Methods\u201d. Global Journal of Science Frontier Research - A: Physics & Space Science GJSFR-A Volume 22 (GJSFR Volume 22 Issue A3).
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.