Juvenile GM1-gangliosidosis, also known as type II or juvenile GM1-gangliosidosis, is an autosomal recessive lysosomal storage disorder that clinically differs from infantile GM1gangliosidosis in the absence of the characteristic cherry-red patch and hepatosplenomegaly. The disease is characterized by mild skeletal abnormalities and slowly progressing neurodegeneration. Due to the late age of onset and unusual presentation, diagnostic confusion with other ataxic and purely neurological disorders is common. There are currently 3-4 recognized types of GM1-gangliosidosis, with type I being the most prevalent phenotype with an average onset age of 6 months. Several subtypes of GM1-gangliosidosis are caused by mutations in the GLB1 gene, but the location and type of deleterious mutations have a direct impact on the severity of the disease and the age at which it manifests.
## I. INTRODUCTION
For cells to function efficiently, the processes of glycoconjugate production and degradation need to be carefully controlled. Glycoconjugates are essential for the majority of biological processes. galactosidase, also known as GAL, is a lysosomal hydrolase that is responsible for the degradation of a wide variety of glycoconjugates. This is accomplished by hydrolyzing the non-reducing end of glycan moieties. This enzyme's primary role is to delink galactose residues from one another. According to research [1, 2], GM1 ganglioside and its asialo derivative GA1 have a tendency to concentrate in the lysosomes that are present in brain tissue. The clinical signs of the illness are caused by neurodegenerative pathways in the brain that are triggered when there is an excess of ganglioside—Galactosidase substrate. The accumulation of GM1 gangliosides in microglial cells of the central nervous system has been demonstrated to result in greater activation and infiltration of inflammatory cells into these cells, according to studies conducted using animal models. Previous research [3] has shown that inflammation seems to have a key role in both the etiology of the disorder as well as the neurological symptoms of the condition. It is believed that GM1 gangliosidosis affects between 1 in 100,000 and 200,000 neonates [4]. These numbers are based on estimates from previous studies. Type II GM1 gangliosidosis, sometimes called juvenile or late infantile GM1 gangliosidosis, is distinguished by the slow onset and progression of clinical signs. Ataxia is often the first obvious symptom associated with this subtype, followed by dystonia and spasticity. People who have type II GM 1 gangliosidosis do not have the usual signs of hepatosplenomegaly, cherry red patches, or distinctive facial characteristics. This makes it challenging to make an accurate diagnosis of the condition. People who have this syndrome seem to develop normally in the early stages of the illness. However, symptoms often begin to manifest between the ages of 3 and 5 in those affected by the juvenile form, but they appear sooner in those affected by the late infantile variety. The clinical appearance of GM1 gangliosidosis type II is characterized by diminished neurodevelopmental abilities, including motor and verbal skills. This is one of the disease's defining characteristics. Those who are affected may also have seizures that are difficult to control, which is another potential symptom. Previous research [5] has uncovered conclusions that are comparable to this one. Although the patient had an atypical clinical appearance, it was more suggestive of Zellweger syndrome than anything else. Zellweger syndrome is a hereditary condition that may be identified by the presence of peroxisome deficits. Hypotonia, often known as a loss of muscular tone, and weak or nonexistent vocalizations are two of the hallmarks of this condition, which is frequently brought on by mutations in the PEX gene. Infants affected by this disorder often struggle to feed and may experience the development of seizures at an earlier age.
## II. ETHICAL APPROVAL
The patient's mother consented to the publication of this deidentified case report. Institutional review board approval is not required for deidentified single case reports or histories based on institutional policies.
## III. CLINICAL SUMMARY
We describe a case of an 8-month-old baby who was identified as having Type 2 GM-1 Gangliosidosis. After an uneventful first pregnancy, the patient was the third child of the consanguineous, healthy parents. His older sister appears to be completely normal. The patient had an inguinal hernia, which was discovered during the prenatal ultrasound screening. The patient was once sent to the hospital at the age of 2 months for a hernia operation, during which it was discovered that he had breathing problems. However, with proper measures the surgical procedure was conducted and the patient was shifted to the ICU for a day. Gradually the patient became better with continuous nebulization and was finally discharged. At 8 months of age, the patient's parents again reported to the hospital with complaints of difficulty in breathing, periorbital puffiness and fever since 3 days in the child. The mother also noticed that the baby was having difficulty in sucking milk and drinking and used to intermittently stop feeding. An increased incident of sweating was observed in the baby while feeding. Upon taking the history, it was revealed that the baby had a running nose and history of cough at 5 months of age for which he had taken treatment from a pulmonologist. Upon examination, it was found that the infant showed clinical signs of pneumonia, bilateral hydrocele, macrocephaly, dolichocephaly, frontal bossing, hypotonia, rickets, and global developmental delay [FIGURE 1] A Zellweger syndrome suspect was identified.
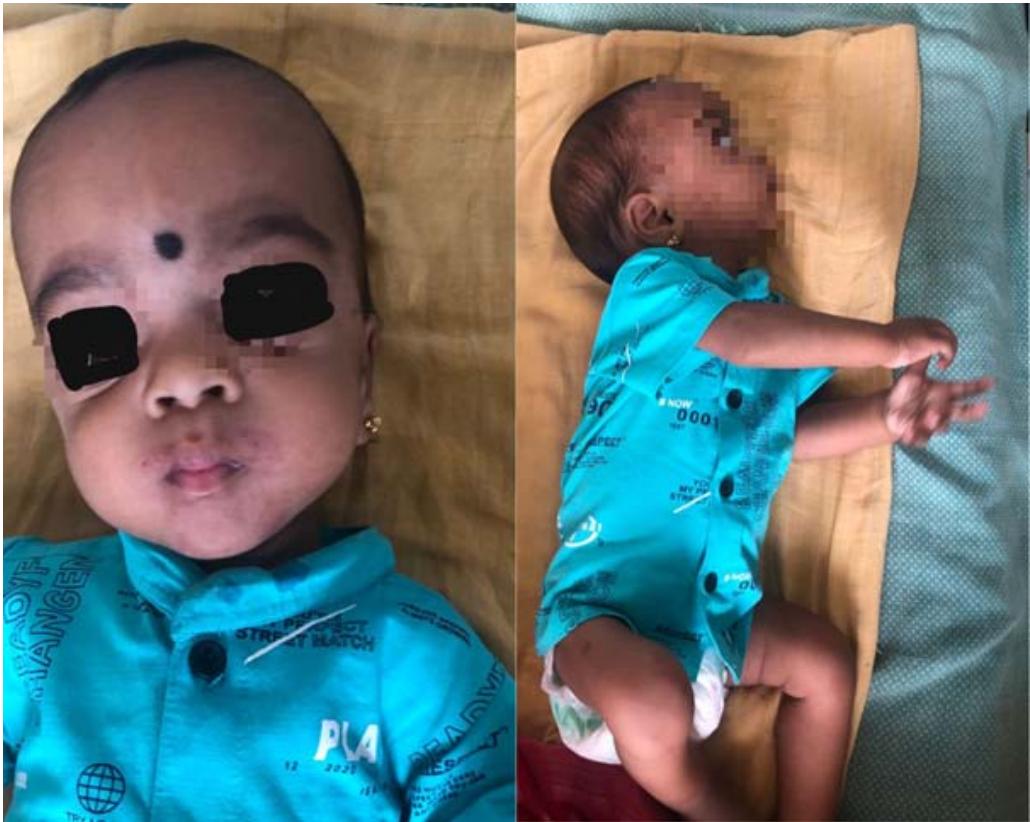 Morphological features of the face and exhibition of hypotonia
The infant was found to have bilateral enlarged kidneys and hepatosplenomegaly upon abdominal examination. There was also a slight ascites present. The brain's magnetic resonance imaging (MRI) revealed widespread corpus callosum thinning, moderately dilated bilateral occipital horns, and insufficient myelination in parieto-occipital white matter. The infant screened positive for rickets, bicytopenia, and severe anaemia in the lab. He had stage 2 hypotension, a well-functioning dilated left ventricle, mild pericardial effusion, and bilateral pleural effusion, according to his echocardiography. The patient's symptoms were controlled while a confirmative diagnosis was made through gene testing.
A homozygous single base pair deletion in exon 10 of the GLB1 gene, which causes a frameshift and an early truncation of the protein 11 amino acids downstream to codon 327, was discovered, according to the gene report. Another homozygous 2-base pair deletion in exon 11 of the CEP41 gene was discovered
[FIGURE 2], which causes a frameshift and an early truncation of the protein downstream of codon 346. The mutation in the CEP41 gene may be significant, but the gene testing reports classify it as a variant of unknown importance because it is placed in the gene's last exon and its impact on protein alteration cannot be predicted. There was a dearth of literature supporting this variety.
Gene transcripts showing various variations ADDITIONAL FINDINGS: VARIANT OF UNCERTAIN SIGNIFICANCE (VUS)
<table><tr><td>Gene# (Transcript)</td><td>Location</td><td>Variant</td><td>Zygosity</td></tr><tr><td>GLB1 (-)
(ENST00000307363.1 0)</td><td>Exon 10</td><td>c.979del
(p.Gln327SerfsTer1 1)</td><td>Homozygous</td></tr></table>
<table><tr><td>Gene# (Transcript)</td><td>Location</td><td>Variant</td><td>Zygosity</td></tr><tr><td>CEP41 (-)(ENST00000675138.1)</td><td>Exon 11</td><td>c.1036_1037del(p.Asn346LeufsTer?)</td><td>Homozygous</td></tr></table>
<table><tr><td>Gene Transcript</td><td>Location</td><td>Variation</td><td>Zygosity</td></tr><tr><td>GLB1(-)
(ENST00000030763.10)</td><td>Exon 10</td><td>c.979del
(p.Gln327SerfsTer11)</td><td>Homozygous</td></tr></table>
<table><tr><td>Gene Transcript</td><td>Location</td><td>Variation</td><td>Zygosity</td></tr><tr><td>CEP41(-)(ENST00000675138.1)</td><td>Exon 11</td><td>C.1036_1037del(p.Asn346LeufsTer)</td><td>Homozygous</td></tr></table>
## IV. DISCUSSION
In this particular instance, there are a few aspects that should be brought to the forefront. The diagnosis of GM-1 Gangliosidosis Type 2 was arrived at after taking into account the clinical phenotype in addition to specific laboratory and genetic abnormalities. The condition known as juvenile GM1 gangliosidosis, which is passed down in an autosomal recessive manner and results in neurological regression in those who are affected by it, was just described. Patients affected by GM1 gangliosidosis type I begin to display clinical symptoms within the first month of their lives. People who have GM1 gangliosidosis type II continue to reach their typical neurodevelopmental milestones (juvenile form) until late infancy (late infantile form) or late childhood. This is the case even in the juvenile form. Because of this, treatment options for diseases with a later onset, such as enzyme replacement therapy, cell therapy, and bone marrow transplantation, can be more successful if molecular diagnosis is performed early on in pre-symptomatic individuals who have a positive family history. There is currently no simple biochemical test available that can be used for carrier screening in high risk people and their families [6]. It has been reported in the past that patients with type II diabetes have an enzyme activity level that is affected less severely [7]. The GM1 gangliosidosis type II and the discovered mutation in the GLB1 gene appear to be completely correlated with one another, with 100 percent phenotypic plasticity in individuals who are homozygous for the mutation. In spite of the fact that heterozygous carriers for this mutation do not appear to be suffering from any symptoms of illness, there is a risk that they will pass on the deleterious mutation to their offspring. As a consequence of this, people who have had childhood ataxia and the relatives of patients who are already well-known should get a GLB1 genetic test before getting married consanguineously. Consanguineous marriage, a family history of deaths with similar symptoms, increasing ataxia, and neurodevelopmental regression are all factors that assist medical professionals in narrowing down the list of possible alternative diagnoses and advising patients on the most appropriate genetic tests.
## V. CONCLUSION
Juvenile GM1 gangliosidosis type II was shown to have an autosomal recessive variant caused by a missense mutation in the GLB1 gene in our patient. The mutation is a rare previously reported pathologic mutation along with the mutation in CEP41 gene. The significance of the later gene's mutation in the illness of the patient is yet to be discovered.
Our findings support a connection between juvenile gangliosidosis type II patients' ataxia and neurodegeneration and the GLB1 gene mutation.
Generating HTML Viewer...
References
7 Cites in Article
Anna Caciotti,Scott Garman,Yadilette Rivera-Colón,Elena Procopio,Serena Catarzi,Lorenzo Ferri,Carmen Guido,Paola Martelli,Rossella Parini,Daniela Antuzzi,Roberta Battini,Michela Sibilio,Alessandro Simonati,Elena Fontana,Alessandro Salviati,Gulcin Akinci,Cristina Cereda,Carlo Dionisi-Vici,Francesca Deodato,Adele D'amico,Alessandra D'azzo,Enrico Bertini,Mirella Filocamo,Maurizio Scarpa,Maja Di Rocco,Cynthia Tifft,Federica Ciani,Serena Gasperini,Elisabetta Pasquini,Renzo Guerrini,Maria Donati,Amelia Morrone (2011). GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings.
J Jarnes Utz,S Kim,K King,R Ziegler,L Schema,E Redtree (2017). Infantile gangliosidoses: mapping a timeline of clinical changes.
M Jeyakumar,R Thomas,E Elliot-Smith,D Smith,A Van Der Spoel,A Azzo,V Perry,T Butters,R Dwek,F Platt (2003). Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis.
Armstrong-Javors A Chu,C (2014). Child neurology: Exaggerated dermal melanocytosis in a hypotonic infant: a harbinger of GM1 gangliosidosis.
Leonhard Wolfe,Robert Senior,N Ng Ying Kin (1974). The Structures of Oligosaccharides Accumulating in the Liver of G1-Gangliosidosis, Type I.
Walter Acosta,Reid Martin,David Radin,Carole Cramer (2016). High-throughput imaging method for direct assessment of GM1 ganglioside levels in mammalian cells.
K Yoshida,A Oshima,M Shimmoto,Y Fukuhara,H Sakuraba,N Yanagisawa,Y Suzuki (1991). Human betagalactosidase gene mutations in GM1gangliosidosis: a common mutation among Japanese adult/chronic cases.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Narendranath Reddy Ganampet. 2026. \u201cA Case of GM 1 Gangliosidosis Type 2 Mimicking Zellweger Syndrome\u201d. Global Journal of Medical Research - A: Neurology & Nervous System GJMR-A Volume 23 (GJMR Volume 23 Issue A3).
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.