We report a rare case of Wilson’s Disease with neurologic features in a 31-year-old man. This disease consists of a disturbance of copper metabolism secondary to a mutation in the gene responsible for encoding the tissue transporter and the enzyme that incorporates the excess element into bile, generating toxic accumulation in the liver, cornea, and central nervous system. According to his wife, the patient had been treated for an unspecified mood disorder. The clinical picture was characterized by depressive mood, anhedonia, and anxiety. He had his first seizure episode on December 3rd, 2021. He progressed with dysarthria, ataxic gait, dystonia of the right-hand flexor muscles, and intermittent urinary incontinence. Marked worsening was observed after the diagnosis of COVID-19 in February 2022. At the clinical evaluation on March 24th, risorius muscle dystonia (risus sardonicus), resting tremor, and Kayser Fleischer rings at slit-lamp examination was also noted.
## I. INTRODUCTION
Wilson's Disease (WD) is a metabolic disorder resulting from biallelic mutations in the ATP7B gene on chromosome $13^{1,2,3}$ of autosomal recessive inheritance[^3], characterized by the toxic accumulation of this element in the liver, cornea, and central nervous system[^4].
Author α Ω: Neurology Specialist, Hospital das Clínicas Samuel Libânio, 777 Comendador Jose Garcia St, Pouso Alegre, Minas Gerais, MG 37550-000, BRA.
Author o: Neurology Specialist, Fellowship in Vascular Neurology, Hospital das Clínicas Samuel Libânio, 777 Comendador Jose Garcia St, Pouso Alegre, Minas Gerais, MG, 37550-000, BRA.
The incidence of these mutations in newborns was estimated at 1:7,000 in Sardinia, Italy and 1.7:100,000 in the Republic of Ireland, in contrast, the prevalence of the disease has been estimated to be between 1:250,000 and 1:300,000 in Sweden and between 1:30,000 and 1:40,000 in other populations[^7].
Copper is an essential cofactor for several enzymes and is present in foodstuffs such as seafood, pulses, and nuts. Its metabolism is dependent on the ATP7B gene, which is responsible for encoding ceruloplasmin, and on the ATPase, which incorporates it into the bile and allows its exteriorization with the feces 10,11
Due to the absence of these mechanisms, copper accumulates in the liver until it spills over into the bloodstream. High levels of cupremia cause disruption of the blood-brain barrier and deposition with a cytotoxic effect in the striatum, globus pallidus, locus coeruleus, substantia nigra, and cerebral cortex $^{4,12}$.
## II. CASE REPORT
A 31-year-old male, mixed race, bricklayer, residing in Paraisópolis, Minas Gerais State, Brazil. History of alcoholism and drug use. Diagnosis of previous unspecified mood disorder and using fluoxetine $40\mathrm{mg/day}$. No other relevant environmental exposures were reported. Report of a male adult family member diagnosed with liver failure of unknown etiology.
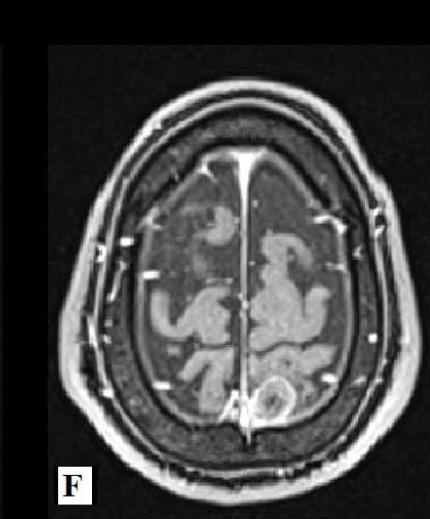
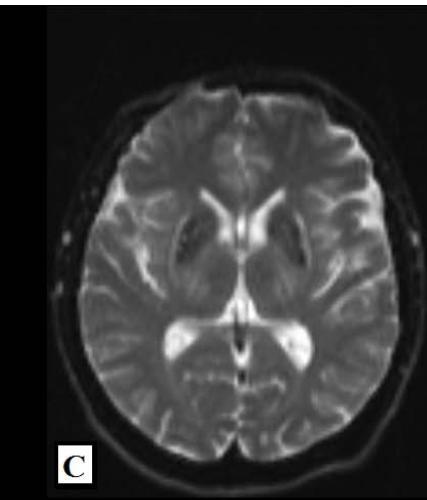
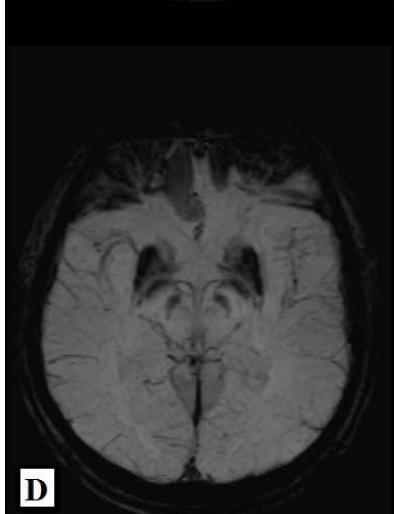
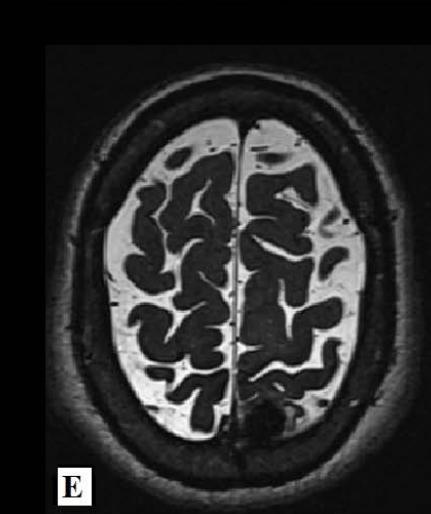
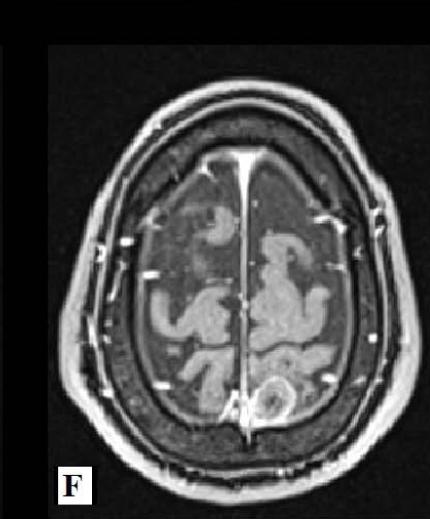
Magnetic Resonance Imaging (MRI) of the brain on December 1st, 2021, showed involvement of the putamen, associated with hemosiderin residue, and crus posterius bilaterally, in addition to the midbrain and pons, without restriction to diffusion (images 1A-1D), and an extra-axial parietal left paramedian contrast-enhanced lesion suggestive of meningioma (images 1E-1F). On December 3rd, 2021, the patient suffered the first generalized clonic tonic seizure while sleeping, and in a follow-up visit on December 21st, he started to use Levetiracetam orally.
He was diagnosed with Covid 19 on February 3rd, 2022, with a mild evolution without the need for ventilatory support or complications. The wife noted that the development of the disease was accentuated after the infection. From February 9th, he appeared to have speech and gait disturbance, difficulty mobilizing the right hand, and urinary incontinence.
On February 16th, he attended the consultation with the responsible physician who, associated the symptoms with the anticonvulsant and switched to Phenytoin $100\mathrm{mg}$ twice a day and associated Dexamethasone orally. It evolved five days later with intermittent hiccups and prostration that lasted approximately three days.
Cerebrospinal fluid (CSF) collected On March 21st revealed a cell count of 0 unit; glucose $89\mathrm{mg / dL}$, lactate $15.7 \, \mathrm{mg/dL}$, gram without staining bacteria, and CSF culture without bacterial growth.
He was hospitalized on March 24th for social reasons to collect WD screening tests. The patient presented to the neurological examination with regular general condition, good spatial orientation, alertness, Glasgow Coma Scale 15, hypomimia, cranial nerve pairs exam without abnormalities, isochoric pupils, normal extrinsic ocular
motricity, risus sardonicus, deep tendon reflexes 2/2, muscle strength 5/5 in all testable upper and lower limb muscle groups, somaesthesia, right-hand flexordystonia, and ataxic gait. Upon slit lamp examination, the presence of Kayser-Fleischer rings was noted.
Laboratory workup of March 24th revealed aminotransferase (AST) 37 mg/dL, alanine aminotransferase (ALT) 35 mg/dL, total bilirubin test 1,00 mg/dL, albumin test 3,7 mg/dL, international normalized ratio (INR) 1,17, and platelets $88 \times 10^{9} / L$. Tests for disorders of Copper metabolism of the same date revealed total serum copper 24,6 mg/dL (reference range (RR) 70mg/dL-150mg/dL), serum ceruloplasmin 7,0 mg/dL (RR 20mg/dL-60mg/dL) and, finally, 24-hours urine copper test 187,4 mg/dL (RR 70mg/dL-150mg/dL). Child-Pugh, Fibrosis-4, and APRI scores were, respectively, 5 points (Child Class A - least severe liver disease); 1,99 (undetermined), and 1,11 (significant fibrosis most likely, cirrhosis undetermined). Abdominal ultrasound exam of April 8th, 2022, indicates chronic liver disease with signs of portal hypertension, splenomegaly, and moderate ascites. Electroneuromyography of March 14th, 2022, was absent abnormalities.
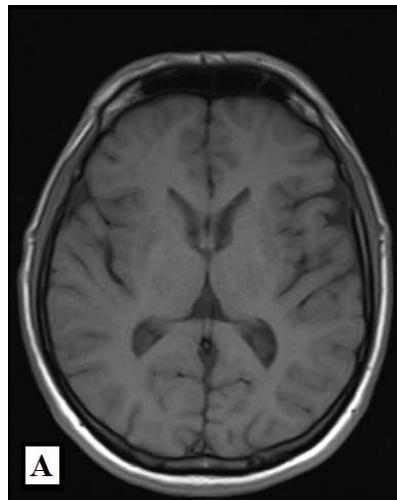
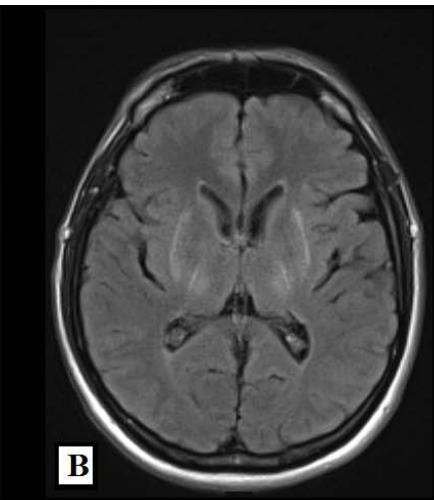
During outpatient follow-up, a new MRI of the brain was requested on April 1st, 2022, which denoted better characterization of foci of signal alteration in cerebellar peduncles (images 2).
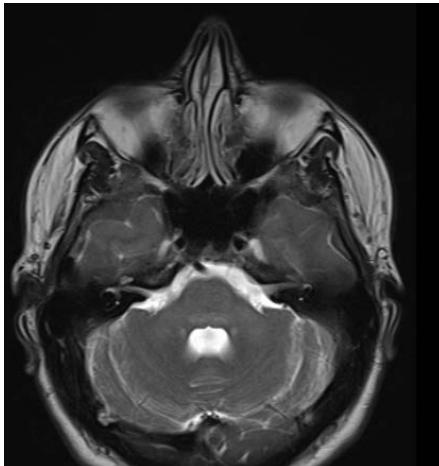 Panel label: A.
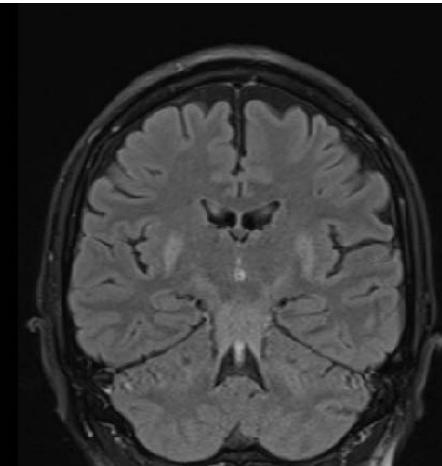 Panel label: B.
On June 2022, he had moderate dysarthria, hypomimia, right-hand flexor dystonia, tetraparesis, bradykinesia, and postural instability, but without rigid or resting tremors.
The specific treatment was started on May 2022 with pyridoxine chlorhydrate $50\mathrm{mg}$ daily and zinc sulfate heptahydrate $4\mathrm{mg/mL}$ 15mL three times a day orally. Due to the cost of the drug, the patient delayed starting penicillamine.
## III. DISCUSSION
Incipient neurological symptoms are subtle and nonspecific, such as difficulty concentrating and motor coordination and handwriting changes (for example, micrograph) $^{13-19}$ and begin on average between 20 and 40 years $^{20-22}$. As it progresses, more prominent symptoms appear, whose order of incidence is dysarthria $(57.6\%)$, dystonia $(42.4\%)$, abnormal gait $(37.8\%)$, tremor $(36.2\%)$, parkinsonism $(17.3\%)$, choreoathetosis $(15.3\%)$ and convulsion $(4.7\%)$ $^{16,17}$. Neurological impairment occurs about a decade after liver failure and, therefore, signs of advanced disease $^{23}$. Cognitive impairment is considered rare and was reported by Machado, Chien, Deguti, et al. $(2006)^{16}$ in $4.2\%$ of cases.
Given the heterogeneity of clinical manifestations, the neurological phenotype of WD can be grouped for didactic purposes into dystonic, pseudosclerotic, parkinsonian, and hyperkinetic subtypes. The patient discussed in this study had a predominance of the dystonic subtype manifested by multifocal dystonia affecting both the risorius muscle (sardonic laughter) and the flexor muscles of the right hand fingers. As reported by Lorincz (2010), bilateral putaminal lesions were found on an MRI of the brain.
Dysarthria can result from any condition that damages the motor control structures necessary for speech production, such as cranial nerves IX, X, XII, cerebellum, and basal ganglia $^{12}$. In this case, it was noted evident bilateral impairment of the basal ganglia.
Seizures are not uncommon and are reported variably in $4.7\%$ 16,17 to $14.5\%$ 25 of WD cases. The patient in question presented, at the initial manifestation, a single episode of generalized tonic-clonic seizure without recurrence.
We also detected the presence of brownish Kayser Fleischer rings, more evident in the lower region of the iris bilaterally. Such a semiological sign is due to copper deposition in the Descemet's membrane of the cornea $^{26}$ and is present in approximately $100\%$ of neuropsychiatric WD cases $^{10}$.
Psychiatric symptoms are reported by about $30\%$ to $60\%$ of individuals affected by WD [26]. In this case, the disorder for which the patient had been using Fluoxetine was not specified. However, the familiar states that at the time of initiation of therapy, he had a depressive mood, anhedonia, and anxiety.
It is possible that such symptoms were already an incipient manifestation of central nervous system involvement.
Cognitive impairment is initially mild and recognized only by family members. It is categorized into frontal lobe syndrome, which involves impulsivity, promiscuity, apathy, hypotenacy, impaired social judgment, planning dysfunction, and emotional lability, and subcortical dementia characterized by slowed thinking amnesia, and executive dysfunction, but without aphasia, apraxia, or agnosia $^{10}$. In this case, it was impossible to attribute a clinical syndrome related to the metabolic disorder, given the history of alcoholism and use of narcotics.
## IV. CONCLUSION
This case report represents the importance of a detailed neurological clinical evaluation and the association of findings with Imaging and laboratory workup. It is a rare disease whose epidemiology in Brazil lacks data, and complementary tests have reduced specificity.
Disclosure statement
No potential conflict of interest was reported by the authors.
Generating HTML Viewer...
References
26 Cites in Article
P Bull,G Tomás,J Rommens,J Forbes,D Cox (1993). The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. Nature Genet 1993;5:327–337.
R Tanzi,K Petrukhin,E Chernov,J Pellequer,W Wasco,B Ross (1993). The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene.
M Faoucher,Caroline (2019). The psychopharmacology of Wilson disease and other metabolic disorders.
Peter Hedera (2019). Wilson's disease: A master of disguise.
G Loudianos,V Dessi,M Lovicu,A Angius,A Figus,F Lilliu (1999). Molecular characterization of Wilson disease in the Sardinian population-evidence of a founder effect.
M Reilly,L Daly,M Hutchinson (1993). An epidemiological study of Wilson's disease in the Republic of Ireland..
C Olsson,E Waldenström,K Westermark,U Landegre,A Syvanen (2000). Determination of the frequencies of ten allelic variants of the Wilson disease gene (ATP7B), in pooled DNA samples.
Louise Azevedo Lobo- Lopes,Sebastião Eurico De Melo- Souza,Vicente Filho,Marco Borges (2021). Mielopathy related to copper deficiency after bariatric surgery: case report / Mielopatia relacionada à deficiência de cobre após cirurgia bariátrica: relato de caso.
Eve Roberts,Michael Schilsky (2008). Diagnosis and treatment of Wilson disease: An update.
M Lorincz (2018). Wilson disease and related copper disorders.
Iury Tussolini,Gabrielle Tussolini,João Tussolini,Luciano Pontes,Mario Rocha Junior,Patrícia Da Silva (2023). Doença de Menkes: relato de um caso raro na Amazônia.
Juan Ortiz,Álvaro Morillo Cox,Willians Tambo,Noha Eskander,Martín Wirth,Margarita Valdez,Maria Niño (2020). Neurological Manifestations of Wilson's Disease: Pathophysiology and Localization of Each Component.
J Walshe (1962). Wilson's Disease: The Presenting Symptoms.
T Saito (1987). Presenting symptoms and natural history of Wilson disease.
Walter Oder,Georg Grimm,Harald Kollegger,Peter Ferenci,Barbara Schneider,Lüder Deecke (1991). Neurological and neuropsychiatric spectrum of Wilson's disease: a prospective study of 45 cases.
Alexandre Machado,Hsin Fen Chien,Marta Mitiko Deguti,Eduardo Cançado,Raymundo Soares Azevedo,Milberto Scaff,Egberto Reis Barbosa (2006). Neurological manifestations in Wilson's disease: Report of 119 cases.
James Burke,Praveen Dayalu,Bin Nan,Fred Askari,George Brewer,Matthew Lorincz (2011). Prognostic significance of neurologic examination findings in Wilson disease.
Anna Członkowska,Tomasz Litwin,Karolina Dzieżyc,Michal Karliński,Johan Bring,Carl Bjartmar (2018). Characteristics of a newly diagnosed Polish cohort of patients with neurological manifestations of Wilson disease evaluated with the Unified Wilson’s Disease Rating Scale.
S Rubinstein,A Young,K Kluin,G Hill,A Aisen,T Gabrielsen (1987). Clinical assessment of 31 patients with Wilson's disease. Correlations with structural changes on magnetic resonance imaging.
Edmund Cauza,Theresia Maier-Dobersberger,Claudia Polli,Klaus Kaserer,Ludwig Kramer,Peter Ferenci (1997). Screening for Wilson's disease in patients with liver diseases by serum ceruloplasmin.
Xueying Xu,Sokhon Pin,Muraya Gathinji,Ralph Fuchs,Z Harris (2004). Aceruloplasminemia: An Inherited Neurodegenerative Disease with Impairment of Iron Homeostasis.
Zeynep Tümer (2013). An Overview and Update of<i>ATP7A</i>Mutations Leading to Menkes Disease and Occipital Horn Syndrome.
Peter Ferenci (2004). Pathophysiology and Clinical Features of Wilson Disease.
Matthew Lorincz (2010). Neurologic Wilson's disease.
L Prashanth,S Sinha,A Taly,A.Mahadevan,M Vasudev,S Shankar (2010). Spectrum of epilepsy in Wilson's disease with electroencephalographic, MR imaging and pathological correlates.
Wolfgang Stremmel,Uta Merle,Ralf Weiskirchen (2019). Clinical features of Wilson disease.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Laryssa Garcia de Almeida. 2026. \u201cNeurological Wilson Disease in a Young Brazilian Adult: A Case Report\u201d. Global Journal of Medical Research - A: Neurology & Nervous System GJMR-A Volume 23 (GJMR Volume 23 Issue A3).
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.