## I. CASE REPORT
### a) Introduction
IgG4-related disease (IgG4-RD), also called IgG4 syndrome or IgG4-associated disease, is a chronic inflammatory condition of presumed autoimmune etiology. It is characterized by the potential for IgG4+ plasma cell infiltration in various tissues and can affect many organs, although the incidence and prevalence have not been established1. Classically, it is described in men — 3 to 4 men for every woman — on average in the sixth decade of life, although rare cases have been identified in other age groups, including children.[2,3]
The disease was first described in the pancreas and was called lymphoplasmacytic pancreatic sclerosis4. In addition to the pancreas, other sites have common involvement such as lymph nodes, bile duct, salivary gland, parotid gland, submandibular gland, lacrimal gland, kidney, retroperitoneum and lung.[1,5]
The prognosis of patients with IgG4-related disease tends to be good after treatment, with mortality being an unexpected event. Complications and functional organ failure due to fibrosis are related to delays in diagnosing the disease.
This disease has been better understood in recent years and, as a potential cause of morbidity, it must be known by the doctor so that there is clinical suspicion, correct diagnosis and appropriate management. In this context, the aim of this paper is to report a case of IgG4-associated disease with cutaneous and lymph node involvement, and to discuss it on the basis of the literature.
### b) Clinical Case
A 53-year-old female patient came for medical assessment complaining of severe pain, difficulty mobilizing her right upper limb and lymphadenomegaly in the right axillary region with phlogistic signs for 10 days. Associated with her current condition, she reported an unmeasured fever, hyporexia and a loss of approximately $4\mathrm{kg}$ over the period.
He said that in the last two years he had seen a sudden increase in several lymph nodes, mainly in the cervical, axillary and inguinal chains, which had regressed after taking antibiotics and corticosteroids. However, she said that the current condition was the most intense she had experienced so far.
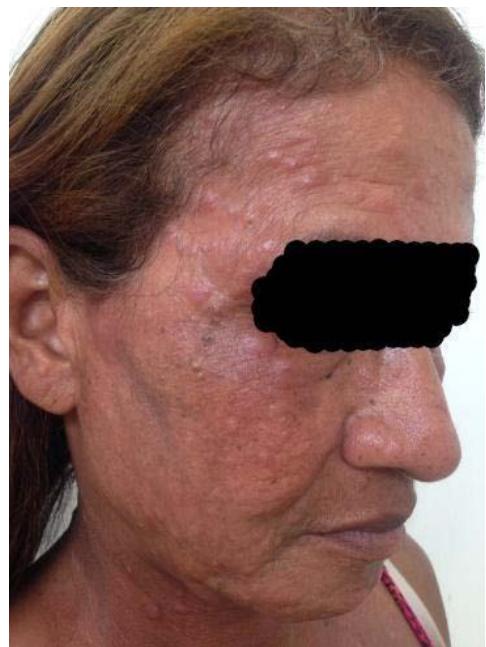
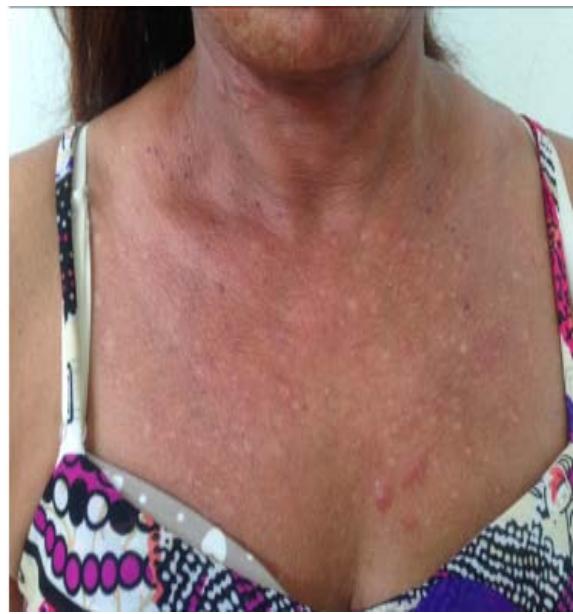
He also reported skin lesions mainly on the face, trunk, and upper limbs for 15 years. He reported that the lesions were erythematous, papulo-nodular and pruritic, which often evolved with ulceration, scarring, and residual dyschromia. Previous biopsies of the skin lesions suggested a diagnosis of rosacea, polymorphous eruption in the light and nodular prurigo. Noteworthy, in her history, she reported treatment for American Tegumentary Leishmaniasis with skin involvement 2 years ago and reported having systemic arterial hypertension.
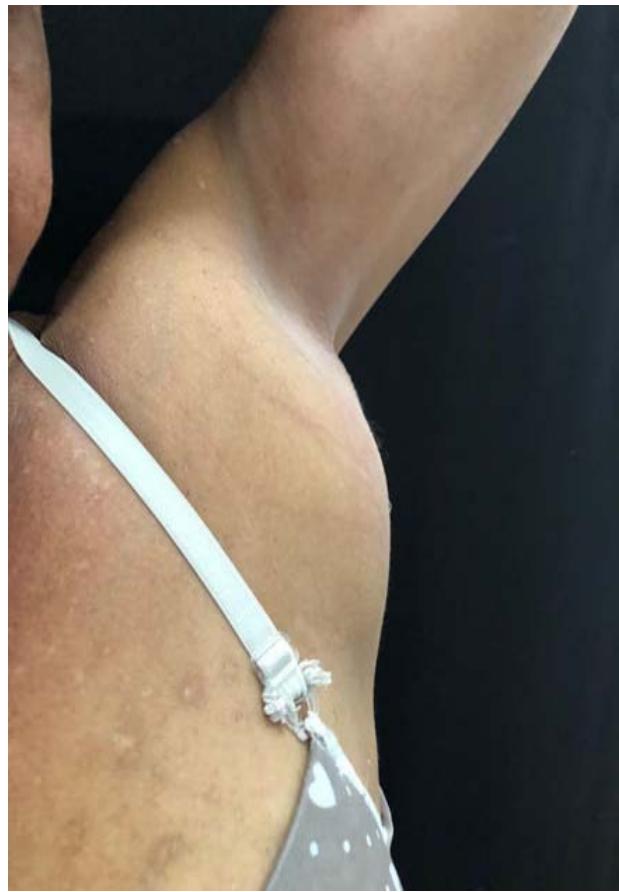
Physical examination in the right axillary region revealed multiple enlarged and confluent lymph nodes, forming a mass of approximately $5\mathrm{cm}$ in its largest diameter. Presence of associated heat, redness, and edema. Impression of mobile and fibroelastic lymph nodes varying in size from approximately 1 to $3\mathrm{cm}$ without associated phlogistic signs, palpable in the left axillary, cervical and inguinal regions bilaterally. In addition, erythematous papulo-nodular lesions were present mainly on the face and neck, and to a lesser extent on the neck, trunk and limbs, the latter also showing a marked presence of residual dyschromia.
The patient underwent tests for diagnostic investigation, considering the hypotheses of lymphoproliferative neoplasia, ganglionic tuberculosis, histoplasmosis and paracoccidioidomycosis.
Computed tomography of the chest showed the presence of confluent lymphadenomegaly in the right axillary chain, forming a mass with a center of necrotic, measuring approximately $56 \times 40 \mathrm{~mm}$ in their largest diameters. Other non-confluent lymph nodes of increased size were observed in the ipsilateral axillary chain, left axillary chain and retroperitoneum. Abdominal CT scans showed prominent lymph nodes in the common iliac chain and bilateral inguinal nodes.
The following were negative: blood culture; culture for mycobacteria and fungi in lymph node aspirates; serology for paracoccidioidomycosis, histoplasmosis and coinfections; non-reactive PPD.
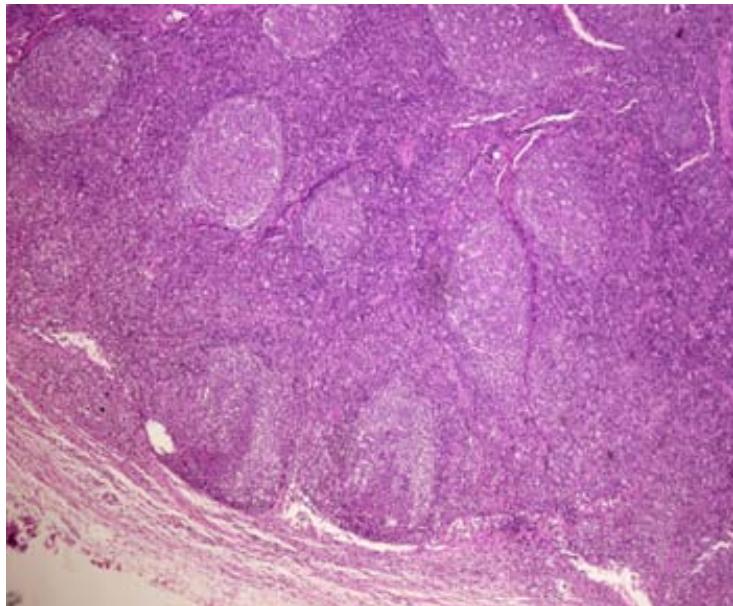
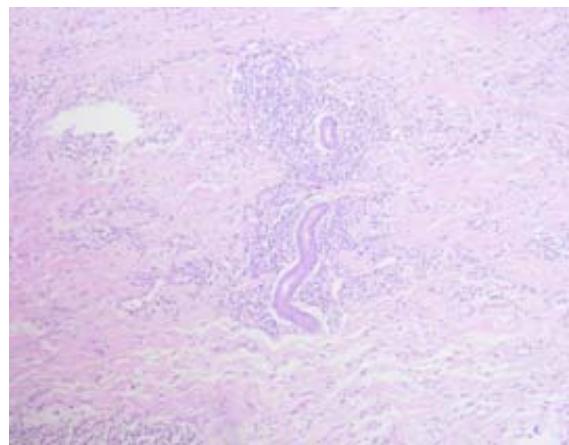
The skin biopsy of the right axillary region showed epidermis with pseudoepitheliomatous acanthosis, a dense inflammatory infiltrate made up of lymphocytes, plasma cells and histiocytes, permeated by some neutrophils and rare osinophils in the dermis. No parasites were found in the skin fragment evaluated. An excisional biopsy of the right inguinal lymph node showed reactive lymphadenopathy with a pattern of follicular and paracortical hyperplasia, with no evidence of neoplasia.
Other complementary tests were requested, including total IgG 2130 mg/dL; IgG4 562 mg/dL; rheumatoid factor 28.8 UI/ml; and C4: 5.75 mg/dl; FAN 1:640 homogeneous pattern; anti Sm negative; anti-Dna negative; anti-SSA negative; anti SSb: negative.
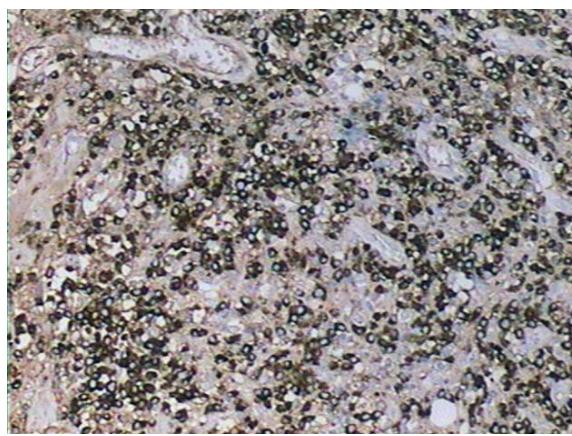
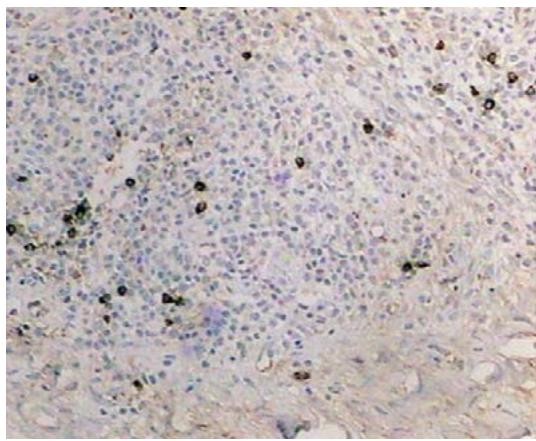
Based on the patient's history of recurrent lymphadenomegaly, the presence of papulo-nodular lesions with a chronic course, a skin biopsy with lymphoplasmocytic infiltrate and an increase in IgG4, the hypothesis of a disease was put forward related to IgG4. Immunohistochemistry of the previous biopsy taken from the right axillary region was requested. The immunohistochemistry was positive for CD138, a plasma cell antigen. There were more than 30 IgG4 cells per high magnification field, which corroborated the diagnosis of IgG4-related disease with lymph node and skin involvement.
In this context, to induce remission, the decision was made to introduce corticosteroids at a dose of 40 mg/day of prednisone, with a good clinical response and initial regression of the lymphadenopathy and skin lesions. However, when the steroid dose was reduced, the patient's skin lesions and lymphadenopathy tended to recur, preventing complete withdrawal. Therefore, two cycles of Rituximab 1,000 mg were carried out with a 15-day interval between them, with the aim to avoid the side effects of prolonged corticosteroid use. The patient progressed with an excellent response to the immunobiologic. The corticosteroid was completely withdrawn. Rituximab was maintained every 6 months for control.
### c) Discussion
The pathogenesis of IgG4-related disease is not yet fully understood, but it is presumed to be an autoimmune disease. Some autoantigens have already been proposed, such as galectin 3, laminin 111 and annexin A11, but further studies need to be carried out to better confirm this6-8. The CD4 + cytotoxic T lymphocyte appears to be of fundamental importance in the pathogenesis, being found in large numbers scattered among the lesions of the affected organs9. Through the production of granzyme B, perforin, IL1, TGF beta and gamma interferon, CD4 + cytotoxic T cells appear to act on the fibrosis often present in this disease10. Another cell that has aired importance in the pathogenesis are T Helper follicular (Thf) cells, which help in the differentiation of B cells. Thf cells are increased in affected tissue and in peripheral blood, the concentration of which is related to disease activity $^{11}$.
Currently, it is postulated that the TH2 response, previously considered the main pathway in the etiology of IgG4 syndrome, plays a marginal role in the pathogenesis of the disease and is seen more frequently in atopic patients12. It should be noted that IgG4 syndrome has been better understood and studied recently, so it is believed that new discoveries regarding pathogenesis will occur over time.
It is described in the literature that IgG4 syndrome, in addition to organ involvement, can present non-specific subacute symptoms and influence the patient's general condition5. Furthermore, it usually involves two or more organs, which can be impacted synchronously or metachronously1. The skin is not a site commonly involved in IgG syndrome4. Skin lesions were found in $4.2\%$ of patients in a study of 118 Chinese affected by the disease and in $6.3\%$ in a study of 80 Japanese patients13,14 In the case described, the affected sites were the skin and lymph nodes, in addition to nonspecific symptoms such as malaise and loss of weight were present, characterizing involvement beyond the specific organ.
Lymphadenopathy is commonly present in IgG-related disease4 and can be generalized or localized. Generally, the lymph nodes do not grow more than 2 cm nd patients remain afebrile $^{15}$. Lymph node involvement is typically observed together with other clinical findings of the syndrome but may be the initial or only manifestation of the syndrome $^{16}$. The mediastinal, hilar, intra-abdominal and axillary chains are the main sites of lymph node involvement, and multiple lymph node chains are usually impacted $^{17}$.
The clinical picture of skin involvement is poorly described compared to that of other classically affected organs. However, when present, skin lesions present as nodules, papules or plaques which may be pruritic, preferentially affecting the head and neck and, less commonly, the trunk and extremities $^{15}$. Skin involvement is strongly associated with lymph nodes -in $46\%$ of cases-; lacrimal glands -in $33.3\%$; orbit -in $30\%$; and salivary glands -in $53\%$. Autoimmune pancreatitis, on the other hand, is less common than is usually found in cases with cutaneous involvement $^{18}$.
Other differential diagnoses include skin lesions that can mimic nodular prurigo, rosacea, angiolymphoid hyperplasia with eosinophilia, xanthogranulomas and cutaneous lymphomas19. Lymph node enlargement is often confused with lymphoma, sarcoidosis, multicentric Castleman's disease or other malignancies5. Therefore, a correct diagnosis is essential, excluding malignancies, so that the patient can have better clinical management and avoid unnecessary treatment.
In the case described, for both the cutaneous and lymph node involvement, different diagnostic hypotheses were previously proposed until the correct diagnosis was reached. The skin lesions had previously been diagnosed as nodular prurigo, polymorphous light eruption and rosacea, which is part of the differential diagnosis reported in the literature. On the other hand, in the differential diagnosis of lymph node involvement, the lymphoproliferative neoplasms have been suggested, as well as infectious diseases with lymph node involvement. Against this backdrop, it is important to know more about the syndrome IgG4 so that it can be better understood, enabling clinical suspicion and early diagnosis.
In 2019, the American College of Rheumatology submitted a classification and criteria for the diagnosis of IgG4-related disease. Until then, the proposed criteria were based on three pillars: 1) characteristic clinical criterion of swelling or mass with involvement of one or more organs; 2) hematological criterion with presence of serum IgG4 $>135\mathrm{mg / dL}$; 3) histological criterion with presence of lymphoplasmocytic infiltrate and fibrosis added to a ratio of IgG4+/igG+ plasma cells greater than $40\%$, in addition to the visualization of more than 10 IgG4+ plasma cells per high magnification field18. The presence of criteria 1, 2 and 3 confirms the diagnosis, the presence of criteria 1 and 3 makes the case probable and the presence of criteria 1 and 2 makes the case possible. Therefore, it should be emphasized that an increase in serum IgG4 levels is not necessary or specific for the diagnosis 20, 21. Although histopathology is considered by some authors to be the gold standard test, it should not be evaluated in isolation from the context of a patient with clinical signs suggestive of IgG4- associated disease.[21] In skin or lymph node biopsies, you don't necessarily find all the classic histopathological findings. The expected findings described in the literature include a dense lymphoplasmocytic infiltrate rich in IgG4, obliterative phlebitis and storiform fibrosis. However, these findings can vary according to the organ affected and the patient. Histopathology of the skin usually shows a lymphoplasmacytic infiltrate in the dermis and/or subcutaneous tissue, with perivascular and perianexial involvement. Obliterative phlebitis and a storiform pattern of fibrosis are rarely present. The lymph nodes also often don't show the clinical signs described, such as storiform fibrosis 15,21.
In the case of the patient under study, the skin and lymph node biopsies did not show the 3 classic findings. The lymph node biopsy was able to rule out evidence of malignancy and the infectious diseases considered. The histological criteria were better met with a skin biopsy of the right axillary region, which showed a significant lymphoplasmocytic infiltrate. Subsequently, this biopsy Immunohistochemistry was carried out on the skin, which revealed positivity for CD138 plasma cell antigen and more than 30 IgG4+ cells per high magnification field.
Imaging tests can help to corroborate clinical and histopathological suspicions, elucidate the involvement of the disease, and are useful for better choosing biopsy sites and helping with patient follow-up. These tests include ultrasound (USG), computed tomography (CT), magnetic resonance imaging (MRI) and positron emission tomography (PET-CT) $^{5,22}$. As it is a disease with the potential to affect several organs, the active search for other affected sites should be encouraged. In the case studied, the CT scan was of great importance as it revealed the involvement of numerous lymph node chains, although the presence of the disease in organs other than the skin and lymph nodes was not found.
In IgG4-associated disease, some complementary tests may be found to be positive more often than in the general population. These include those that are frequently positive — in more than $50\%$ of cases — which can be increased IgG4, IgG or IgE; those that are commonly positive — found in 25 to $50\%$ of cases — such as FAN, rheumatoid factor and complement drop; and those that are uncommon — in less than $25\%$ of cases, such as increased CRP[1]. In the case under study, the patient had an increase in serum IgG and IgG4, a FAN pattern of 1/640 (homogeneous) and a drop in C4, which is in line with what has been reported in the literature without this meaning another disease.
Other studies defend the importance of plasma blasts, which are an oligoclonal population of CD19+ CD20- CD27+ CD38+, precursors of tissue antibodies. Plasma cells are increased in active disease, which can be seen using flow cytometry. For some authors, the plasma blast count correlates with the activity of the disease and, with the When treatment is started, the value decreases and, in the presence of relapses, the value increases more reliably than IgG4 levels[4,5]. The plasma blast count, however, is not universally available in practice and, for the same reason, could not be carried out on the patient reported.
The criteria proposed by the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) in 2019 establish the need for involvement of an organ typically affected in the syndrome, exclusion of other etiologies for the case and laboratory evidence of IgG4-RD (serological, histological, imaging)[27]. Our patient had lymph node and skin involvement. As already mentioned, the skin is not a frequently affected organ. On the other hand, lymph node involvement is frequent, as is skin involvement associated with lymph node involvement $^{18,28}$.
With regard to treatment, a consensus supports the idea that systemic corticosteroids, when indicated, are the first line of therapy for inducing remission. Generally, the equivalent prednisone dose of 0.6 mg/kg/day is given initially, and lower doses can be administered in maintenance therapy to prevent relapses when necessary. Azathioprine (2 mg/kg/day), Mycophenolate (up to 2.5g/day) and other immune-suppressants appear as second-line treatment with the aim of maintaining remission and avoiding the damage secondary to prolonged steroid use $^{5,15}$. Although corticosteroid sparing agents are frequently used in cases of autoimmune pancreatitis and sclerosing cholangitis related to IgG4 syndrome, more studies need to be carried out to better evaluate the efficacy of these medications $^{23,24,25}$.
Rituximab (anti-CD20) has recently been reported as a promising treatment for IgG4-associated disease, probably acting on plasma blasts, reducing IgG4 production and disease activity $^{5,15}$. An intravenous dose of 1 g every 15 days for a total of 2 doses is the dose initially suggested for Rituximab, which has been shown to be effective both in inducing remission and in maintenance $^{26}$.
## II. CONCLUSION
Based on the theoretical arguments discussed, it can be concluded that this was a case of IgG4-related disease that manifested itself clinically with involvement skin and lymph nodes. The diagnosis was made by combining clinical, histopathology, immunohistochemistry and other findings in blood and imaging tests. As this entity has only recently been described in the literature, much remains to be discovered about the pathogenesis, management and long-term outcome of patients with this disease. Therefore, further studies are needed to gain a better understanding of IgG4- related disease.
With this case, we emphasize that in the case of papulonodular lesions and large masses of adenomegaly, skin biopsy can be a valuable and easily accessible tool for diagnosing IgG4 syndrome, ruling out neoplasms, avoiding inappropriate management or a late diagnosis.
## III. ATTACHMENTS
 Panel label: A.
 Panel label: C.
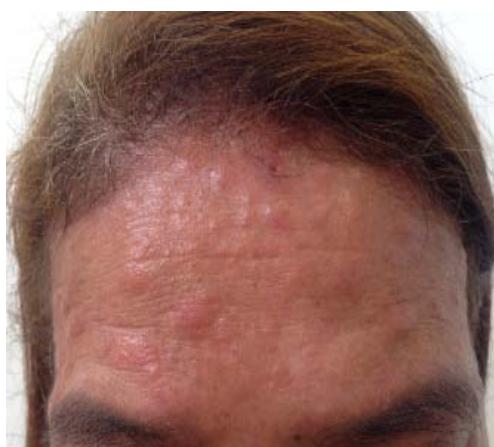 Panel label: B.
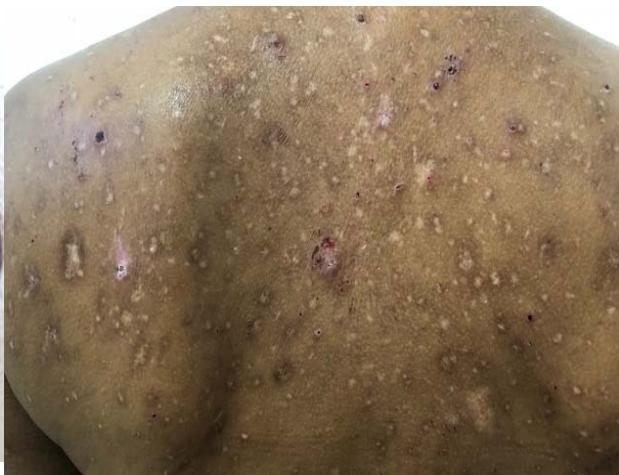 D Fig. 1: (A and B) Presence of erythematous papulo-nodular lesions affecting the face and cervical region; (C) Presence of papulo-nodular lesions affecting the neck and evidence of post inflammatory hypochromia lesions in the same topography; (D) Predominance of residual dyschromia on the back.
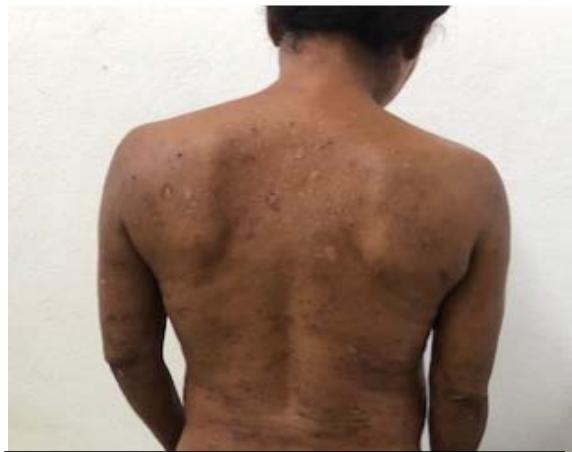

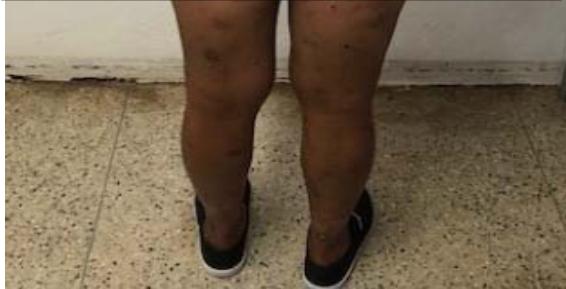 Panel label: A.
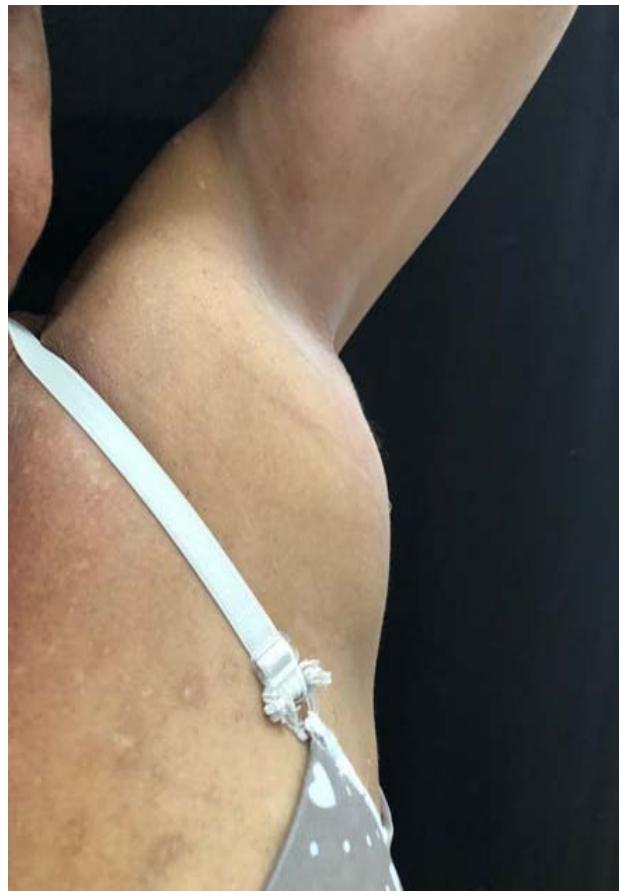 Panel label: B.
 C Fig. 2: (A) Extent of skin involvement with a predominance of residual dyschromias; (B) Evidence of lymphadenomegaly with mass effect in the left axillary chain; (C) Lymph node biopsy site in the right inguinal chain.
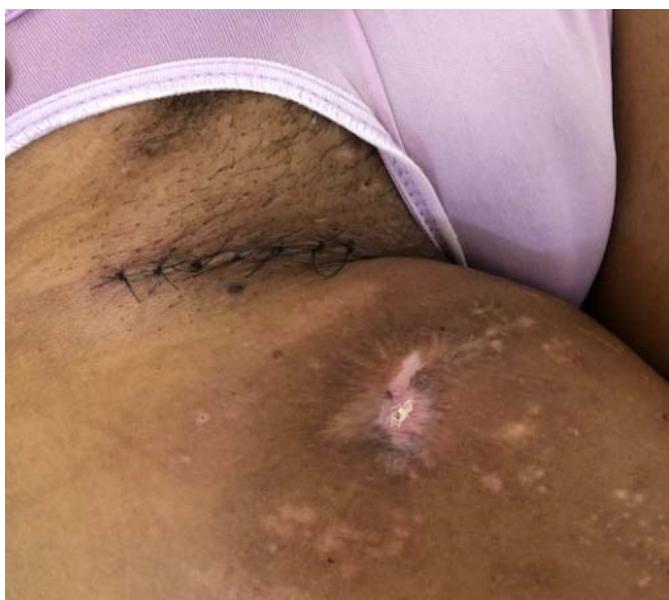
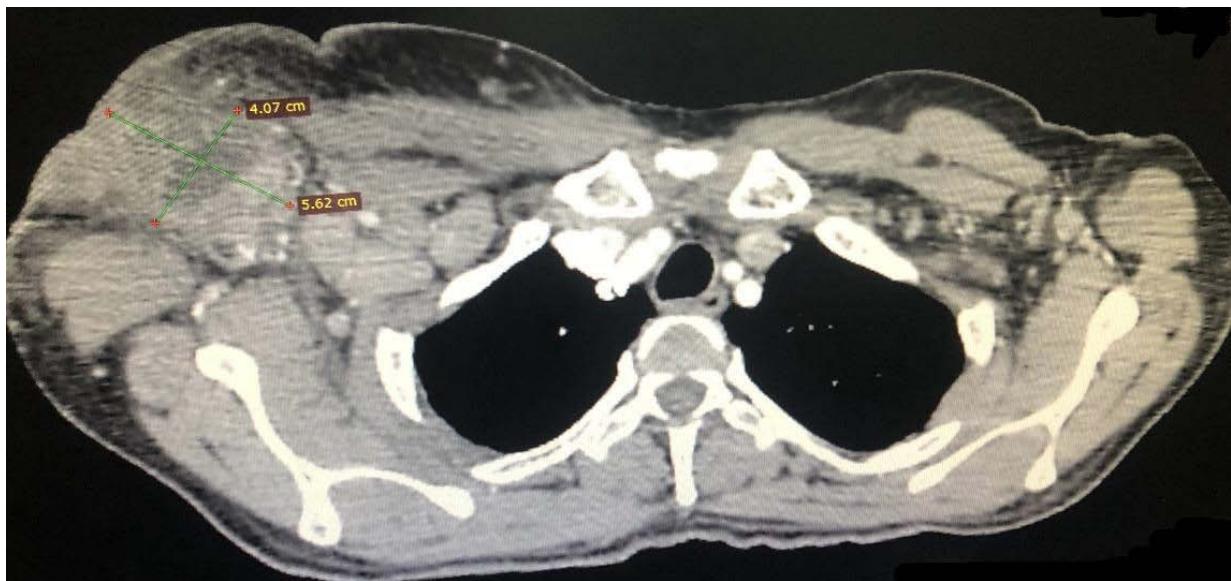 Panel label: A.
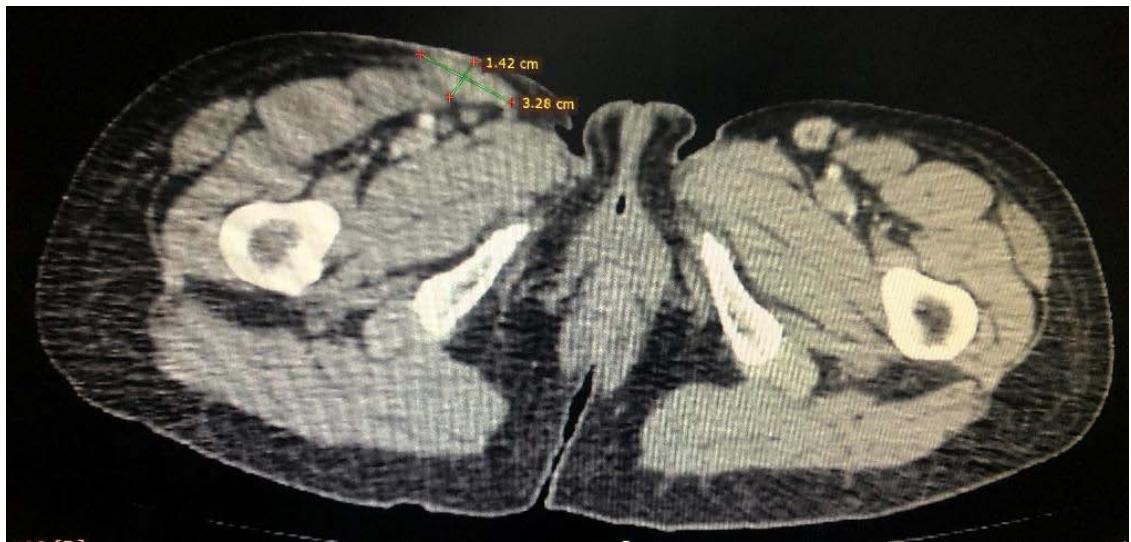 Panel label: B.
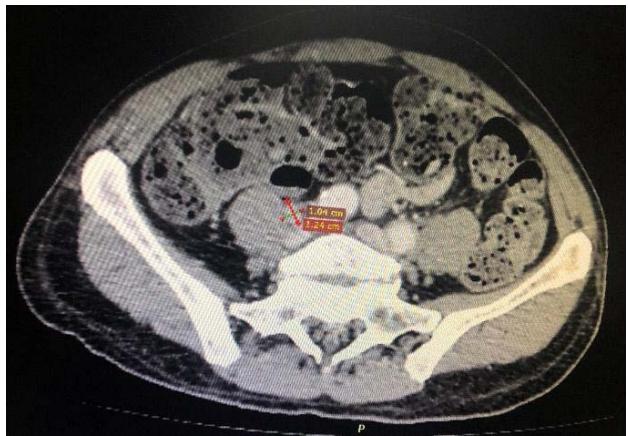 C Fig. 3: (A) CT scan of the right axillary region showing confluent lymph nodes forming a mass with a necrotic center measuring approximately $56 \times 40 \mathrm{~mm}$ in its largest diameters; (B and C) CT scan showing lymphadenopathy in chains in the right inguinal region and the right common iliac region respectively.

 Panel label: AB.
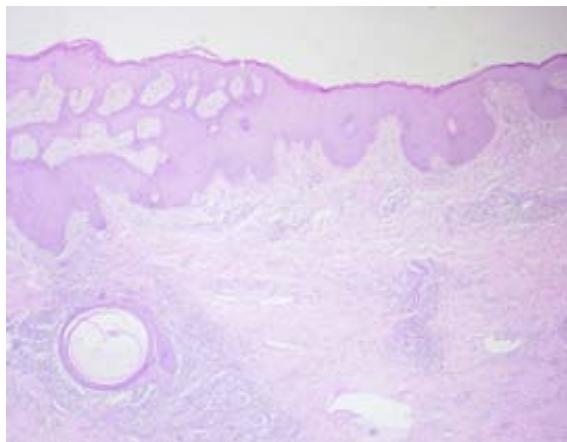 Fig. 4: Reactive lymphadenopathy with a pattern of follicular and paracortical hyperplasia, with no evidence of neoplasia
 Panel label: B. Panel label: A.
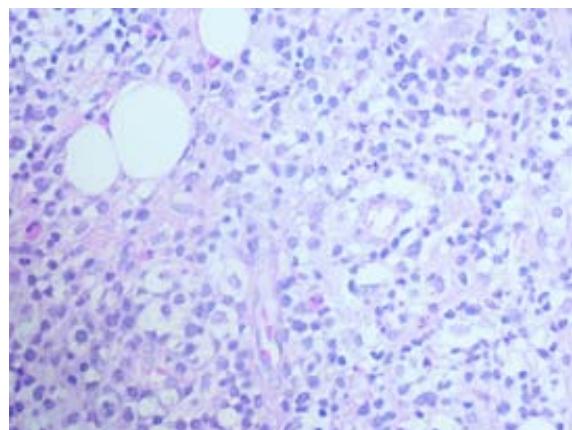 C Fig. 5: (A) Lower magnification showing epidermis with irregular pseudoepitheliomatous acanthosis; (B and C) Dense inflammatory infiltrate made up predominantly of lymphocytes, plasma cells and histiocytes, permeated by some neutrophils and rare eosinophils, with no signs of parasites
 Panel label: A.
 Panel label: B.
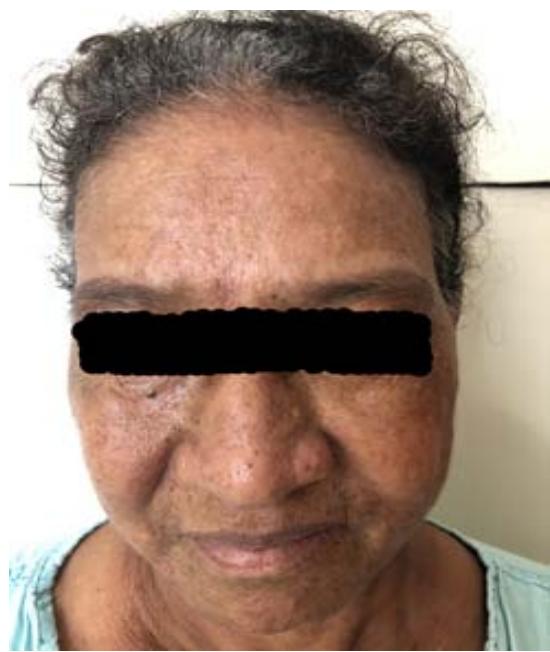 Fig. 6: (A) Immunohistochemistry positive for CD138 in plasma cells (Clone M115); (B) Immunohistochemistry focally positive for IgG4 immunoglobulin (Clone HP6025) - 30 IgG4+ cells per high magnification field A
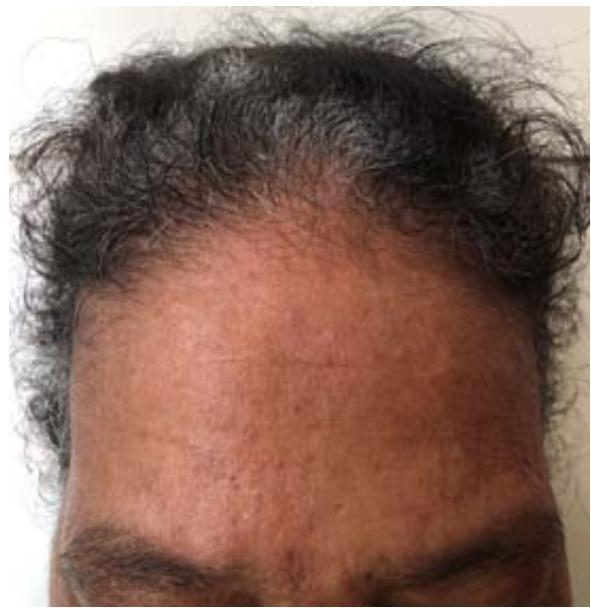 Panel label: B.
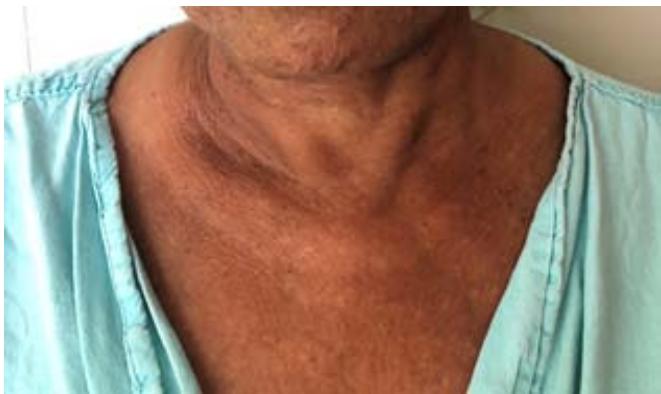 C Fig. 7: (A, B and C) Remission of IgG4 syndrome-related skin lesions during prednisone maintenance at a dose of 40mg/day (0.6mg/kg/day)
Generating HTML Viewer...
References
29 Cites in Article
K Miyabe,Y Zen,Cornell Lyn (2018). Gastrointestinal and Extra-Intestinal Manifestations of IgG4-Related Disease.
A Codina,F Valle,P Blanca (2015). IgG4-Related Disease: Results from a Multicenter Spanish Registry.
Mmf Nastri,G Novak,Aem Sallum (2018). Immunoglobulin G4-related disease with recurrent uveitis and kidney tumor mimicking childhood polyarteritis nodosa.
E Cassione,J Stone (2017). IgG4-Related Disease.
A Wolfson,D Hamilos (1000). Recent advances in understanding and managing IgG4-related disease.
C Perugino,S Alsalem,H Mattoo (2019). Identification of galectin-3 as an autoantigen in patients with IgG4-related disease.
M Shiokawa,Y Kodama,K Sekiguchi (2018). Laminin 511 is a target antigen in autoimmune pancreatitis.
Lowiek Hubers,Harmjan Vos,Alex Schuurman,Robin Erken,Ronald Oude Elferink,Boudewijn Burgering,Stan Van De Graaf,Ulrich Beuers (2018). Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease.
Hamid Mattoo,Vinay Mahajan,Takashi Maehara,Vikram Deshpande,Emanuel Della-Torre,Zachary Wallace,Maria Kulikova,Jefte Drijvers,Joe Daccache,Mollie Carruthers,Flavia Castelino,James Stone,John Stone,Shiv Pillai (2016). Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease.
E Della-Torre,E Bozzalla-Cassione,C Sciorati (2018). A CD8α-Subset of CD4+SLAMF7+ Cytotoxic T Cells Is Expanded in Patients With IgG4-Related Disease and Decreases Following Glucocorticoid Treatment.
Y Chen,Lin Yang,H (2018). Aberrant Expansion and Function of Follicular Helper T Cell Subsets in IgG4-Related Disease.
H Mattoo,E Della-Torre,V Mahajan,J Stone,S Pillai (2014). Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy.
Kazunori Yamada,Yasuhito Hamaguchi,Takako Saeki,Kunimasa Yagi,Naoko Ito,Yasushi Kakuchi,Masakazu Yamagishi,Kazuhiko Takehara,Yasuni Nakanuma,Mitsuhiro Kawano (2013). Investigations of IgG4-related disease involving the skin.
Adele Shenoy,Nirupa Mohandas,Alice Gottlieb (2019). Cutaneous and systemic IgG4-related disease: a review for dermatologists.
W Cheuk,H Yuen,S Chu (2008). Lymphadenopathy of IgG4-related sclerosing disease.
H Saegusa,M Momose,S Kawa (2003). Hilar and pancreatic gallium-67 accumulation is characteristic feature of autoimmune pancreatitis.
Adam Bennett,Neil Fenske,Paul Rodriguez‐waitkus,Jane Messina (2016). IgG4‐related skin disease may have distinct systemic manifestations: a systematic review.
Garrett Lowe,Ryan Bogner,Rokea El‐azhary,Tania Gonzalez‐santiago,Scott Kindle,Julia Lehman,Lawrence Gibson (2015). Cutaneous manifestations of immunoglobulin <scp>G</scp>4‐related disease: what dermatologists need to know.
Phil Hart,Mark Topazian,Thomas Witzig,Jonathan Clain,Ferga Gleeson,Robin Klebig,Michael Levy,Randall Pearson,Bret Petersen,Thomas Smyrk,Aravind Sugumar,Naoki Takahashi,Santhi Vege,Suresh Chari (2013). Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience.
Matthew Huggett,E Culver,M Kumar,J Hurst,M Rodriguez-Justo,M Chapman,G Johnson,S Pereira,R Chapman,George Webster,E Barnes (2014). Type 1 Autoimmune Pancreatitis and IgG4-Related Sclerosing Cholangitis Is Associated With Extrapancreatic Organ Failure, Malignancy, and Mortality in a Prospective UK Cohort.
H Sekigychi,R Horie,M Kanai (2016). IgG4-Related Disease: Retrospective Analysis of One Hundred Sixty-Six Patients.
Mikael Ebbo,Aurélie Grados,Maxime Samson,Matthieu Groh,Anderson Loundou,Aude Rigolet,Benjamin Terrier,Constance Guillaud,Clarisse Carra-Dallière,Frédéric Renou,Agnieszka Pozdzik,Pierre Labauge,Sylvain Palat,Jean-Marie Berthelot,Jean-Loup Pennaforte,Alain Wynckel,Céline Lebas,Noémie Le Gouellec,Thomas Quémeneur,Karine Dahan,Franck Carbonnel,Gaëlle Leroux,Antoinette Perlat,Alexis Mathian,Patrice Cacoub,Eric Hachulla,Nathalie Costedoat-Chalumeau,Jean-Robert Harlé,Nicolas Schleinitz (2017). Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients.
Z Wallace,R Naden,S Chari,H Choi,E Della-Torre,J Dicaire,P Hart,D Inoue,M Kawano,A Khosroshahi,K Kubota,M Lanzillotta,K Okazaki,C Perugino,A Sharma,T Saeki,H Sekiguchi,N Schleinitz,J Stone,N Takahashi,H Umehara,G Webster,Y Zen,J Stone (2019). American College of Rheumatology/European League Against Rheumatism IgG4-Related Disease Classification Criteria Working Group. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease.
G Katz,J Stone (2021). Papulonodular Lesions in Photo Exposed Areas as First Manifestation of IgG4-Related Disease.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Araújo FM. 2026. \u201cPapulonodular Lesions in Photoexposed Areas as the First Manifestation of IgG4-Related Disease\u201d. Global Journal of Medical Research - F: Diseases GJMR-F Volume 24 (GJMR Volume 24 Issue F2).
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.