Abstrack-Introduction: High-throughput sequencing facilitates the diagnosis of Usher syndrome and other conditions involving deafness and blindness that are genetically related, with improvements not only in accurate diagnosis, time savings, and genotype-phenotype correlation. Advances in genomic sequencing also makes it possible to approach isolated or remotely located populations with community genetics methodologies.
## I. INTRODUCTION
Usher syndrome (USH) is a rare autosomal recessive condition characterized by degenerative vision, sensorineural hearing loss
Author 6: Hospital Departamental San Vicente de Paul - Garzón - Huila - Colombia.
Author 2: School of Medicine, Universidad de Antioquia, Medellin, Colombia.
and, sometimes, vestibular dysfunction [1]. USH is classified into four main types and at least 16 genes have been implicated in their etiology.
High-throughput sequencing (HTS) and whole exome sequencing (WES), are the preferred molecular diagnostic tools for USH diagnosis, as well as for other retinal dystrophies and hearing loss disorders in which genetic heterogeneity is present [2]. HTS has made it possible to identify candidate genes, pathogenic variants, genotype-phenotype correlations, rare undiagnosed diseases and information on the phenotypic spectrum of the disease [2].
This article presents as WES was performed to evaluate and precisely diagnoses a highly inbred family group affected by Usher type 2C syndrome, those who had not been able to be adequately studied, because they are located in a place of Colombia with scarce health resources, near a low complexity hospital, in a context of community genetics.
## II. MATERIALS AND METHODS
### a) Subjects
The index case (III,2) was ascertained by blindness and hearing loss into the hospital departmental San Vicente de Paul de Garzón (Huila), Colombia, the regional hospital facility located from the family's habitual residence. A genealogy was constructed showing that there were other affected members in the place of origin (III,1 deceased, III,4, III,6 and IV,1), including an inbreed union between siblings with an affected child (Figure 1A). A team was then organized with the ophthalmologist, undergraduate medical students with the support from geneticists located in a molecular genetics' laboratory in the capital city of Colombia. The team went to the nursing home and Pitalito (Huila), and evaluate the rest of the affected and healthy relatives. In summary, three more patients were identified, evaluated and sampled, so, they did not have any access to other additional functional tests.
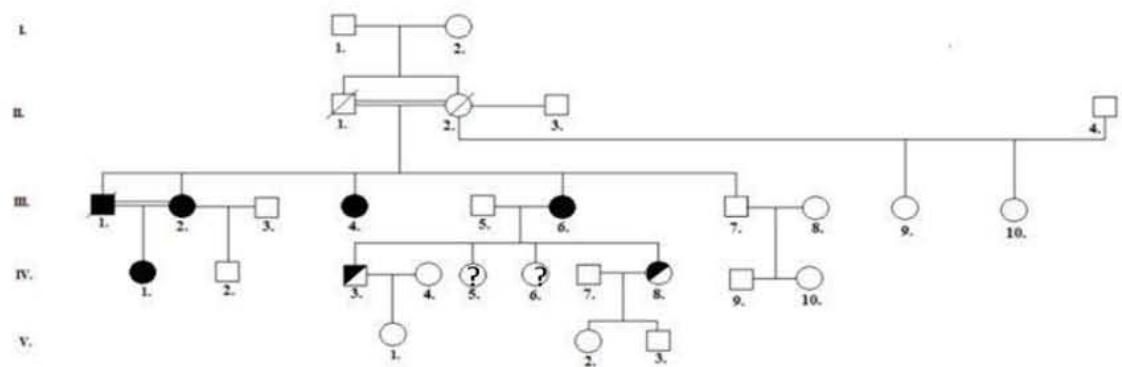 Figure 1A. Genealogical Tree
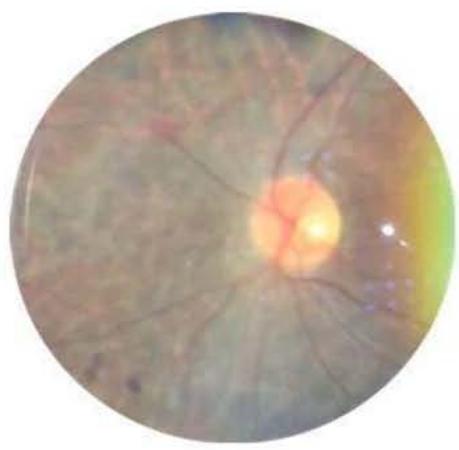 Figure 1B. Index Patient right eye funduscopy
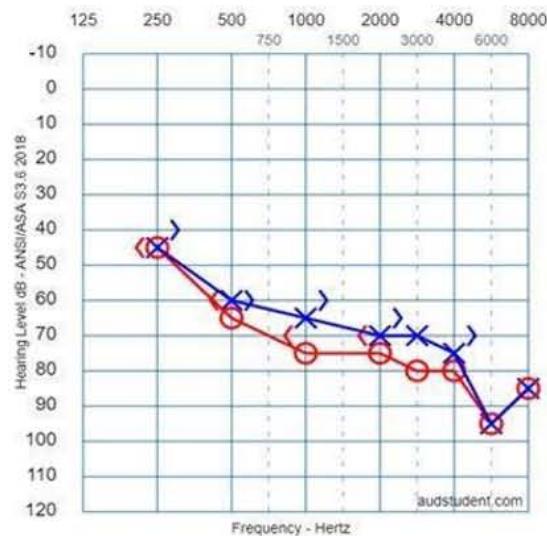 Figure 1C. Index Patient Tonal Audiometry
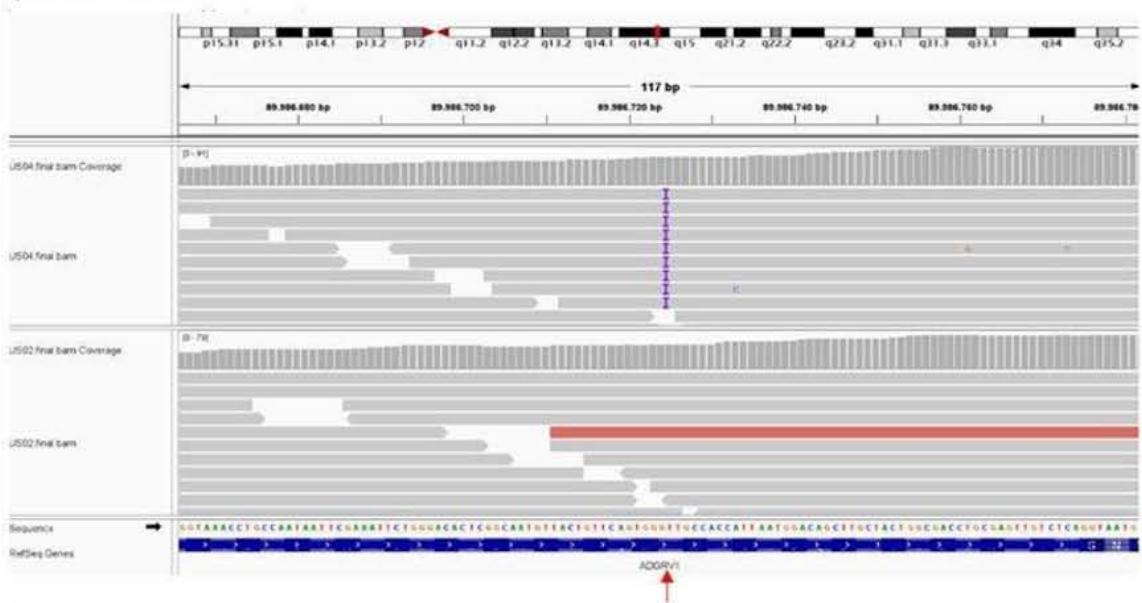 Figure 1D. BAM Archives Figure 1B. Index patient right eye funduscopy. Pink optic disc, with 0.2 excavation, with defined edges, attenuated/retinal vessels of central emergence, speckled pattern with granulation of the retinal pigmented epithelium, presence of bone spicules towards the equator, clear vitreous. Figure 1C. Tonal audiometry of the index case. Right ear: anacusis. Left ear: severe sensorineural hearing loss. Figure 1D. BAM file showing the patient homozygous for the mutant allele (US04) and homozygous for the Wild Type allele (US02).
### b) Diagnostic Tests
All clinical evaluations, procedures and diagnostic tests had the approval and signature of an informed consent, accompanied by a witness. Given the location of those affected, III,2, III,4, III,6 and IV,1, only diagnostic eye fundus tests were performed on (III,2, III,4, III,6 and IV,1), which data was shown (Figure 1B) and/or audiometry on III,2 (Table 1).
Table 1: Main Usher Syndrome Type 2A (USH2A) Clinical Features
<table><tr><td rowspan="2">USH2A
AFFECTED</td><td rowspan="2">AGE</td><td rowspan="2">SEX</td><td colspan="3">USH MAIN CLINICAL FEATURES</td><td rowspan="2">DEBUTING
AGE</td></tr><tr><td>BLINDNESS</td><td>HEARING
LOSS</td><td>VESTIBULAR</td></tr><tr><td>US04 (III-4)</td><td>68</td><td>F</td><td>+</td><td>+</td><td>-</td><td>CHILDHOOD</td></tr><tr><td>US05 (III-6)</td><td>67</td><td>F</td><td>+</td><td>+</td><td>-</td><td>CHILDHOOD</td></tr><tr><td>US06 (IV-1)</td><td>25</td><td>F</td><td>+</td><td>+</td><td>-</td><td>CHILDHOOD -
ADOLESCENCE</td></tr><tr><td>US07 (III-2)</td><td>52</td><td>F</td><td>+</td><td>+</td><td>+</td><td>CHILDHOOD</td></tr></table>
### c) Exome Sequencing and Data Analysis
Genomic DNAs from four affected individuals (US04, US05, US06, US07) and four healthy relatives (US01, US02, US03, US08) was extracted from whole blood using standard procedures. Sequencing libraries were prepared using $1.0~\mu \mathrm{g}$ of genomic DNA per sample and the Agilent SureSelect Human All Exon kit (Agilent Technologies, CA, USA) following manufacturer's recommendations. Briefly, genomic DNA was fragmented by hydrodynamic shearing to generate 180-280 bp fragments. Next, overhang ends were converted into blunt ends and $3^{\prime}$ ends were adenylated. Then, DNA fragments were ligated to adapter oligonucleotides, and these were enriched by PCR. Exon capture was performed by hybridization using biotin labeled probes and purification by the AMPure XP system (Beckman Coulter, Beverly, USA). Finally, captured libraries were enriched in a PCR reaction and index codes were added to each sample. Sequencing was performed in an Illumina HiSeq 2500 equipment generating paired-end reads. Library preparation and sequencing was carried out by Novogene (Beijing, China).
Raw reads were trimmed, and mapped to the human reference genome (GRCh37) using the Burrows-Wheeler Aligner v0.7.8-r455 (BWA) \[3\](doi: 10.1093/bioin formatics/btp698). Duplicate reads were marked in BAM files with Picard v1.111 and variant calling was performed using the GATK v3.8 \[4\](doi: 10.1101/gr.107524.110). The percentage of reads mapped to the reference genome for all the samples was above $99.9\%$ and the fraction of the targeted region covered with at least 10X was above $95\%$, for all samples. Variant call format (VCF) files were annotated and analyzed using the VarSeq v2.2.3 software. Variants were filtered using a multigene panel: ADGRV1, ARSG, CDH23, CIB2, CLRN1, CRTAC1, ESPN, HARS1, MYO7A, PCDH15, PDZD7, USH1C, USH1G, USH2A and WHRN; minimum allele frequency $(\mathsf{MAF}) < 0.01$ according to gnomAD genome and exome variant frequencies v2.1 [5](doi:10.1038/s41586-020-03174-8); protein effect, including missense, indels, frameshift and splicing site variants; and family segregation, assuming an autosomal recessive inheritance model.
## III. RESULTS
The pedigree identifies five members who manifested USH, two of them were identified as healthy carriers of the mutation for the ADGRV1 gene (Figure 1A). Four of the patients were interviewed to identify the clinical signs as shown in Table 1.
WES showed a new pathogenic variant which was identified in those affected with USH in ADGRV1 NM_032119.4 c.6819dupT gene in homozygosis, which is a new mutation for this gene and it causes an early stop codon NP_115495p.Ala2274Cysfs\*4, that coincides with the pattern of familial segregation in those affected (Figure 1C).
There were no other pathogenic or likely pathogenic variants identified in ADGRV1 and other Usher known genes; in addition, no modifier alleles were identified in other genes in the whole exome sequencing analysis. Five homozygous ADGRV1 NM_032119.4 c.6819dupT NP_115495p.Ala2274Cysfs\*4 affected patients in the present family showed the same phenotype with blindness and hearing loss, even though there were limitations of resources for the detailed visual and hearing diagnosis, for which only two affected patients were tested because of their isolated geographic settlement.
## IV. DISCUSSION
Usher syndrome (USH) is a clinically and genetically heterogeneous hereditary condition which is classified into four different main types, USH1, USH2, USH3 and USH4, depending on the age of onset, severity, progression of the symptoms and the presence or absence of vestibular dysfunction [1]. USH also presents genetic heterogeneity with at least 16 genes involved and further into different subtypes: USH1 is caused by the mutation of six different genes: MYO7A, USH1C, CDH23, PCDH15, USH1G and CIB2; USH2 is caused by the mutation of five genes USH2A, ADGRV1,
WHRN, PDZD7 and DFNB31; USH3 is related with mutation of two genes, CLRN1 and HARS and USH4 is caused by mutations in ARSG gene. In addition, ABHD12 gene is related with hearing loss, retinitis pigmentosa, cataracts and ataxia; CEP78 and CEP250 genes mutations causes two forms of cone-rod dystrophy with hearing loss [6]. This genetic heterogeneity implies difficulties in obtaining an accurate diagnosis in USH and also in no syndromic hearing loss and no syndromic retinitis pigmentosa forms [6], and the adequate correlation between the genotype and the phenotype. USH also has digenic, biallelic and polygenic inheritance, which adds difficulty to establish the precise diagnosis [6].
USH and other related conditions and their clinical and genomic heterogeneity requires a multidisciplinary team supported by complex laboratory equipment at fixed laboratory facilities, and sophisticated analysis to establish a precise diagnosis, to understand genotype-phenotype correlations and other particular issues for a rare disease. Genomic medicine helps us to shortcut those questions and WES analysis is ideal in cases with wide clinical and genetic variability, particularly in cases with real or relative geographical isolation or when it is impossible or difficult to evaluate several patients at the same time and collect all data and diagnostic tests of a sensory type (ophthalmological and auditory). In the present inbred familial case, genomic analysis by means of WES in a community context, through the taking of blood samples from those affected and relatives, quickly allowed obtaining the accurate diagnosis of USH2C in a short period of time, highlighting how DNA sequencing technologies are simplifying and accelerating genetic diagnosis.
USH type 2 (USH2) is the most frequent subtype and to date, three causative genes have been identified: USH2A, WHRN and ADGRV1, respectively causing USH2A, USH2B, USH2C and USH2D. The. ADGRV1 gene has mutations in 6 to $19\%$ of subtype 2 cases [7].
In the present study, a highly inbred family with five affected individuals of both genders with USH2C was identified: nine people were clinically examined: four women were affected, with three of them presenting progressive sensorineural hearing loss and retinitis pigmentosa; one, in addition, presented vestibular alterations evidenced by vertigo and gait impairment (Table 1), with which the diagnosis of USH2C was proposed. In addition, attention was paid to a first degree inbred union in which the parents and the daughter showed homozygosity for the ADGRV1 and USH2C gene mutation with USH2C. Directed exome sequencing study with an Usher syndrome panel rapidly showed a new pathogenic variant in the ADGRV1 gene, confirming not only the accurate diagnosis of the Usher Syndrome subtype, but also the number affected for a family in a small village of Colombia; a fact that surely as a result of geographic isolation, facilitated the intrafamily unions observed in the genealogy.
The ADGRV1 gene codifies an adhesion protein that bines to the V1 receptor of the G- protein which is part of the superfamily of polypeptides coupled to G-protein and expressed in the central nervous system. The ADGRV1 protein functions as a transmembrane receptor with seven domains whose function is conducting the normal stereociliary development in the ear [8]. In the eye, the protein is involved in the growth of retinal photoreceptors [7]. Mutations in this gene have been related to four phenotypes: Usher syndrome type 2C with autosomal recessive inheritance, such as the present familial case; Usher syndrome type 2C, along with a polymorphism in the PDZ7 gene in biallelic digenic inheritance, autosomal dominant isolated sensorineural deafness and familial febrile seizure syndrome type 4 of autosomal dominant inheritance [9-12] ADGRV1 NM_032119.4 c.6819dupT gene in homozygosis, which is a new mutation for this gene and it causes an early stop codon NP_115495p. Ala2274Cysfs\*4. WES analysis rapidly showed they differ because in USH type I, hearing loss is congenital and profound, with absence of vestibular functions and progressive retinitis pigmentosa with onset in childhood; USH type II has moderate to severe sensorineural hearing loss and retinitis pigmentosa with onset in the second decade of life; on the other hand, USH type III presents progressive sensorineural hearing loss and retinitis pigmentosa with onset in the second decade of life and variable vestibular dysfunction
Next-generation sequencing identified a new pathogenic variant in the gene ADGRV1 NM_032119.4 c.6819dupT in homozygosity which caused an early stop codon NP_115495 p.Ala2274Cysfs\*4, which predicts the synthesis of a short and possibly aberrant protein that affects the function of the G- protein complex.
In summary, a highly inbred family, located in a remote part of Colombia was identified with a pathogenic variant in the ADGRV1 gene which causes USH2C in a fast way and with few resources of medical infrastructure. In addition, a new mutation was found for this gene. Emphasis is given to the usefulness of next-generation sequencing for a rapid solution and diagnosis of difficult cases.
## V. CONCLUSIONS
Next -generation sequencing is a useful tool to identify mutations and in the rapid and accurate diagnosis of rare diseases such as Usher Syndrome type 2C in remote places, that also was able to be discovered because this family had a highly inbred factor between them, which allowed a new mutation of a recessive syndrome to take place in their genomics.
## ACKNOWLEDGMENTS
The research team thanks the Universidad del Rosario for providing its facilities, equipment, and personnel to carry out all the molecular and genetic analysis of the samples evaluated. Likewise, we thank the Hospital San Vicente de Paul for collaborating with the recruitment of the evaluated subjects. Finally, we thank our patients for allowing us to publicize their case and broaden the knowledge of the disease.
Generating HTML Viewer...
References
12 Cites in Article
Carla Fuster-García,Belén García-Bohórquez,Ana Rodríguez-Muñoz,Elena Aller,Teresa Jaijo,José Millán,Gema García-García (2021). Usher Syndrome: Genetics of a Human Ciliopathy.
I Perea-Romero,F Blanco-Kelly,I Sanchez-Navarro,I Lorda-Sanchez,S Tahsin-Swafiri,A Avila-Fernandez,I Martin-Merida,M Trujillo-Tiebas,R Lopez-Rodriguez,Rodriguez De Alba,M Iancu,I Romero,R Quinodoz,M Hakonarson,H Garcia-Sandova,B Minguez,P Corton,M Rivolta,C Ayuso,C (2021). NGS and phenotypic ontology-based approaches increase the diagnostic yield in syndromic retinal diseases.
Heng Li,Richard Durbin (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform.
Aaron Mckenna,Matthew Hanna,Eric Banks,Andrey Sivachenko,Kristian Cibulskis,Andrew Kernytsky,Kiran Garimella,David Altshuler,Stacey Gabriel,Mark Daly,Mark Depristo (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data.
M Stemerdink,B García-Bohórquez,R Schellens,G Garcia-Garcia,E Van Wijk,J Millan (2021). Genetics, pathogenesis and therapeutic developments for Usher syndrome type 2.
(2022). National Center for Biotechnology Information (NCBI).
D Wu,W Huang,Z Xu,S Li,J Zhang,X Chen,Y Tang,J Qiu,Z Wang,X Duan,L Zhang (2020). Clinical and genetic study of 12 Chinese Han families with nonsyndromic deafness.
Zubin Saihan,Andrew Webster,Linda Luxon,Maria Bitner-Glindzicz (2009). Update on Usher syndrome.
José Millán,Elena Aller,Teresa Jaijo,Fiona Blanco-Kelly,Ascensión Gimenez-Pardo,Carmen Ayuso (2011). An Update on the Genetics of Usher Syndrome.
G Jouret,C Poirsier,M Spodenkiewicz,C Jaquin,E Gouy,C Arndt,M Labrousse,D Gaillard,M Doco-Fenzy,A-S Lebre (2019). Genetics of Usher Syndrome: New Insights From a Meta-analysis.
No ethics committee approval was required for this article type.
Data Availability
Not applicable for this article.
How to Cite This Article
Juan Carlos Kuan. 2026. \u201cWhole Exome Sequencing Identifies a Novel Mutation in ADGRV1 Responsible For Usher 2C Syndrome in a Large Inbred Family.\u201d. Global Journal of Science Frontier Research - G: Bio-Tech & Genetics GJSFR-G Volume 23 (GJSFR Volume 23 Issue G1): .
Explore published articles in an immersive Augmented Reality environment. Our platform converts research papers into interactive 3D books, allowing readers to view and interact with content using AR and VR compatible devices.
Your published article is automatically converted into a realistic 3D book. Flip through pages and read research papers in a more engaging and interactive format.
Abstrack-Introduction: High-throughput sequencing facilitates the diagnosis of Usher syndrome and other conditions involving deafness and blindness that are genetically related, with improvements not only in accurate diagnosis, time savings, and genotype-phenotype correlation. Advances in genomic sequencing also makes it possible to approach isolated or remotely located populations with community genetics methodologies.
Our website is actively being updated, and changes may occur frequently. Please clear your browser cache if needed. For feedback or error reporting, please email [email protected]
Thank you for connecting with us. We will respond to you shortly.